Enzyme

{kind=link}

.svg.png)
Une enzyme est une protéine dotée de propriétés catalytiques. Pratiquement toutes les biomolécules capables de catalyser des réactions chimiques dans les cellules sont des enzymes ; certaines biomolécules catalytiques sont cependant constituées d'ARN et sont donc distinctes des enzymes : ce sont les ribozymes.
Une enzyme agit en abaissant l'énergie d'activation d'une réaction chimique, ce qui accroît la vitesse de réaction. L'enzyme n'est pas modifiée au cours de la réaction. Les molécules initiales sont les substrats de l'enzyme, et les molécules formées à partir de ces substrats sont les produits de la réaction. Presque tous les processus métaboliques de la cellule ont besoin d'enzymes pour se dérouler à une vitesse suffisante pour maintenir la vie. Les enzymes catalysent plus de 5 000 réactions chimiques différentes[2]. L'ensemble des enzymes d'une cellule détermine les voies métaboliques qui peuvent avoir lieu dans cette cellule. L'étude des enzymes est appelée enzymologie.
Les enzymes permettent à des réactions de se produire des millions de fois plus vite qu'en leur absence. Un exemple extrême est l'orotidine-5'-phosphate décarboxylase, qui catalyse en quelques millisecondes une réaction qui prendrait, en son absence, plusieurs millions d'années[3],[4]. Comme tous les catalyseurs, les enzymes ne sont pas modifiées au cours des réactions qu'elles catalysent, et ne modifient pas l'équilibre chimique entre substrats et produits. Les enzymes diffèrent en revanche de la plupart des autres types de catalyseurs par leur très grande spécificité. Cette spécificité découle de leur structure tridimensionnelle. De plus, l'activité d'une enzyme est modulée par diverses autres molécules : un inhibiteur enzymatique est une molécule qui ralentit l'activité d'une enzyme, tandis qu'un activateur de cette enzyme l'accélère ; de nombreux médicaments et poisons sont des inhibiteurs enzymatiques. Par ailleurs, l'activité d'une enzyme décroît rapidement en dehors de sa température et de son pH optimums.
Structure
Les enzymes sont généralement des protéines globulaires qui agissent seules, comme le lysozyme, ou en complexes de plusieurs enzymes ou sous-unités, à l'instar du complexe α-cétoglutarate déshydrogénase. Comme toutes les protéines, les enzymes sont constituées d'une ou plusieurs chaînes polypeptidiques repliées pour former une structure tridimensionnelle correspondant à leur état natif. La séquence en acides aminés de l'enzyme détermine la structure de cette dernière, structure qui, à son tour, détermine les propriétés catalytiques de l'enzyme[5]. Bien que ce soit la structure d'une enzyme qui détermine sa fonction, il n'est pas encore possible à ce jour de prédire l'activité d'une nouvelle enzyme en ne connaissant que sa structure[6]. La structure des enzymes est altérée (dénaturée) lorsqu'elles sont chauffées ou mises en contact avec des dénaturants chimiques, ce qui a généralement pour effet de les inactiver.
Les enzymes sont des molécules bien plus grandes que leurs substrats. Leur taille varie de 62 résidus pour le monomère de 4-oxalocrotonate tautomérase[7] à plus de 2 000 résidus pour l'acide gras synthase animale[8]. Seule une toute petite partie de l'enzyme — entre deux et quatre résidus le plus souvent — est directement impliquée dans la catalyse, ce qu'on appelle le site catalytique. Ce dernier est situé à proximité d'un ou plusieurs sites de liaison, au niveau desquels les substrats sont liés et orientés afin de permettre la catalyse de la réaction chimique. Le site catalytique et les sites de liaison forment le site actif de l'enzyme. Le reste de la protéine sert à maintenir la configuration du site actif et à y générer les conditions optimales pour le déroulement de la réaction.
Dans certains cas, la catalyse ne fait intervenir aucun des résidus d'acides aminés de l'enzyme mais plutôt un cofacteur lié à cette enzyme. La structure des enzymes peut également contenir un site de liaison pour un effecteur allostérique qui provoque un changement conformationnel activant ou inhibant l'activité enzymatique.
Mécanisme
Liaison au substrat
Les enzymes doivent tout d'abord se lier à leurs substrats avant de pouvoir catalyser toute réaction chimique. Les enzymes sont généralement très spécifiques en ce qui concerne à la fois les substrats auxquels elles peuvent se lier et les réactions qu'elles sont susceptibles de catalyser. Cette spécificité résulte de la configuration de leurs sites de liaison, qui sont des poches présentant une complémentarité de forme ainsi que de distribution spatiale des charges électriques et des propriétés hydrophile/hydrophobe par rapport à celles du substrat. Les enzymes peuvent ainsi faire la différence entre des molécules très semblables, ce qui assure leur chimiosélectivité, leur régiosélectivité et leur stéréospécificité[9].

Certaines des enzymes parmi les plus spécifiques interviennent dans la réplication de l'ADN et l'expression génétique. Certaines de ces enzymes sont pourvues d'un mécanisme de « correction d'épreuve » : c'est le cas des ADN polymérases, qui sont capables de corriger leurs erreurs de réplication — paires de bases incorrectes — avant de passer au nucléotide suivant[10]. Ce processus en deux étapes permet d'atteindre une fidélité particulièrement élevée, avec moins d'une erreur sur 100 millions de réactions chez les mammifères. Des mécanismes semblables existent également dans les ARN polymérases[11], les aminoacyl-ARNt synthétases[12] et les ribosomes[13]. A contrario, certaines enzymes présentent une ou plusieurs activités dites « de promiscuité », c'est-à-dire qu'elles peuvent catalyser un ensemble de réactions apparentées sur un ensemble de substrats ayant des implications physiologiques diverses. De nombreuses enzymes possèdent des activités catalytiques mineures qui peuvent se manifester fortuitement et être le point de départ pour la sélection de nouvelles fonctionnalités au cours de l'évolution[14],[15].
Modèle de la serrure et de la clef
Afin d'expliquer la spécificité des enzymes dans la sélections des réactions chimiques qu'elles sont susceptibles de catalyser, le chimiste allemand Emil Fischer proposa en 1894 que l'enzyme et le substrat d'une réaction possèdent une géométrie complémentaire permettant au substrat de s'emboîter exactement dans l'enzyme[16]. Cette représentation est souvent appelée « modèle de la serrure et de la clef ». Ce modèle rend compte de la spécificité des enzymes mais ne permet pas d'expliquer comment les enzymes parviennent à stabiliser l'état de transition au cours des réactions.
Modèle de l'ajustement induit
Le biochimiste américain Daniel Koshland proposa en 1958 le modèle dit de l'ajustement induit (induced fit en anglais) comme adaptation du modèle de la serrure et de la clef pour tenir du compte du fait que les enzymes sont des molécules flexibles : plutôt qu'imaginer l'emboîtement d'un substrat dans une enzyme rigide, Koshland considérait que l'interaction entre le substrat et l'enzyme remodelait en permanence le site actif, et ce tout au long de l'établissement de la liaison[17].
Dans certains cas, comme les glycoside hydrolases, le substrat lui-même change légèrement de forme lorsqu'il se lie au site actif de l'enzyme[18].
Le site actif continue de changer de configuration jusqu'à ce que le substrat soit entièrement lié, et ce n'est qu'alors que la distribution des charges et la géométrie finales peuvent être déterminées.
Catalyse
Les enzymes peuvent accélérer des réactions chimiques de plusieurs façons, mais toutes passent par l'abaissement de l'énergie d'activation, notée ΔG‡ (variation d'enthalpie libre) :
- En stabilisant l'état de transition :
- l'enzyme génère un environment dans lequel la distribution de charges est complémentaire de celles de l'état de transition afin d'abaisser son énergie[19] ;
- En ouvrant un chemin réactionnel alternatif :
- l'enzyme peut réagir temporairement avec le substrat et former un intermédiaire covalent ayant un état de transition de plus basse énergie ;
- En déstabilisant l'état fondamental du substrat :
- par déformation du substrat lié dans leur état de transition afin de réduire l'énergie nécessaire pour atteindre cet état[20] ;
- par une orientation du substrat dans une configuration qui réduit la variation d'entropie de la réaction ; la contribution de ce mécanisme à la catalyse est assez faible[21].
Une enzyme peut recourir à plusieurs de ces mécanismes simultanément. Ainsi, les peptidases telles que la trypsine mettent en œuvre une catalyse covalente par triade catalytique, stabilisent la distribution des charges électriques de l'état de transition à l'aide d'un trou oxyanion, et terminent l'hydrolyse en orientant spécifiquement une molécule d'eau.
Dynamique
Les enzymes ne sont pas des structures rigides et statiques. Elles sont au contraire le siège de tout un ensemble de mouvements internes, qu'il s'agisse du mouvement de résidus d'acides aminés individuels, d'un groupe de résidus formant un élément de la structure secondaire, voire d'un domaine entier de la protéine. Ces mouvements donnent naissance à un ensemble de structures légèrement différentes les une des autres qui sont à l'équilibre en perpétuelle interconversion les unes avec les autres. Différents états conformationnels de l'enzyme peuvent par exemple être associés à différentes phases de son activité chimique. Ainsi, différentes conformations de la dihydrofolate réductase sont associées au cours du cycle catalytique respectivement à la liaison avec le substrat, à la catalyse, à la libération du cofacteur, et enfin à la libération du produit[22].
Régulation allostérique
Les sites de régulation allostérique sont des sites de liaison distincts du site actif qui peuvent interagir avec des molécules de l'environnement cellulaire, appelées dans ce cas effecteurs allostériques. La liaison de ces molécules avec ce site induit un changement conformationnel ou une modification de la dynamique interne de l'enzyme, qui altèrent les propriétés du site actif et modifient par conséquent la vitesse de réaction de l'enzyme[23]. Ces changements peuvent activer ou inhiber des enzymes. Les interactions allostériques avec des métabolites en amont ou en aval d'une voie métabolique à laquelle participe l'enzyme provoquent des boucles de rétrorégulation permettant de moduler l'activité de l'enzyme en fonction de l'intensité du flux de métabolites[24].
Cofacteurs
Définitions
Certaines enzymes n'ont besoin d'aucun composant supplémentaire pour être pleinement actives. D'autres ont au contraire besoin d'interagir avec des espèces chimiques non protéiques, appelées cofacteurs, pour être actives. Ces cofacteurs peuvent être inorganiques tels que des ions métalliques ou cluster fer-soufre, ou bien des composés organiques tels qu'une flavine ou un hème. Les cofacteurs organiques peuvent être des coenzymes, qui sont libérées du site actif de l'enzyme au cours de la réaction, ou des groupes prosthétiques, qui demeurent étroitement liés à l'enzyme. Certains groupes prosthétiques organiques sont liés à leur enzyme par covalence, comme c'est le case de la biotine pour des enzymes telles que la pyruvate carboxylase[26].
L'anhydrase carbonique est un exemple d'enzyme à cofacteur de zinc lié à son site actif[27]. Ces ions ou molécules étroitement liées à l'enzyme se trouvent généralement dans le site actif et interviennent dans la catalyse. Ainsi, on trouve fréquemment une flavine ou un hème dans les réactions d'oxydoréduction
Les enzymes dépourvues du cofacteur qui les rend actives sont appelées apoenzymes, ou apoprotéines. Une enzyme liée à son ou ses cofacteurs est appelée holoenzyme. On appelle également holoenzymes les complexes enzymatiques formés de plusieurs sous-unités pour lesquels toutes les sous-unités requises pour l'activité enzymatique sont présentes ; on emploie souvent ce terme pour les ADN polymérases.
Coenzymes
Les coenzymes sont de petites molécules organiques qui peuvent être liées à l'enzyme de façon assez lâche ou, au contraire, très étroite. Elles transportent des groupes fonctionnels ou des résidus d'une enzyme à une autre. Le NAD+, le NADPH et l'ATP sont des coenzymes. Certaines coenzymes telles que la riboflavine, la thiamine et l'acide folique sont des vitamines, c'est-à-dire des composés qui ne peuvent être synthétisés par l'organisme et doivent être absorbés tels quels par l'alimentation. Parmi les groupes chimiques transportés par des coenzymes, on trouve l'ion hydrure H– transporté par le NADH et le NADPH, le groupe phosphate –OPO32– transporté par l'ATP, le groupe acétyle –COCH3 transporté par la coenzyme A, les groupes aldéhyde –CHO, méthényle –CH= ou méthyle –CH3 portés par l'acide folique, ou encore le groupe méthyle porté par la S-adénosylméthionine (SAM).
Dans la mesure où les coenzymes sont modifiées au cours des réactions chimiques catalysées par les enzymes, il peut être utile de les considérer comme des substrats particuliers partagés par de nombreux types d'enzymes. Ainsi, on connaît plus de 1 000 enzymes utilisant le NAD+ comme coenzyme.
Les coenzymes sont régénérées continuellement et leur concentration est maintenue à un niveau constant dans la cellule. Par exemple, le NADPH est régénéré par la voie des pentoses phosphates et la S-adénosylméthionine est régénérée par la méthionine adénosyltransférase. Cette régénération permanente signifie que de petites quantités de coenzymes peuvent être utilisées très intensivement. Par exemple, le corps humain utilise et régénère son propre poids en ATP chaque jour[28].
Thermodynamique

En l'absence de catalyseur (pointillés), les substrats ont besoin d'une énergie d'activation élevée pour atteindre l'état de transition, qui évolue ensuite vers des produits de plus faible énergie.
En présence d'une enzyme (ligne pleine), les substrats se lient à l'enzyme (ES), qui stabilise l'état de transition (ES‡) en réduisant la quantité d'énergie nécessaire pour le former, et le complexe enzyme-produits (EP) est dissocié.
Comme c'est le cas pour tous les catalyseurs, les enzymes de modifient pas la position de l'équilibre chimique d'une réaction. La présence d'une enzyme a simplement pour conséquence d'accélérer la réaction, qui se déroule dans le même sens. Ainsi, l'anhydrase carbonique, qui catalyse une réaction réversible, agit dans l'un et l'autre sens en fonction de la concentration relative de ses réactifs :
- CO2 + H2O → H2CO3 dans les tissus, où la concentration du CO2 est élevée ;
- H2CO3 → CO2 + H2O dans les poumons, où la concentration du CO2 est faible.
La vitesse de réaction dépend de l'énergie d'activation nécessaire pour atteindre, à partir des substrats, l'état de transition, qui évolue ensuite vers la formation des produits de réaction. Les enzymes accélèrent la vitesse de réaction en abaissant l'énergie d'activation de l'état de transition. Elles commencent par établir une liaison enzyme-substrat (ES) de faible énergie, stabilisent par la suite l'état de transition (ES‡) de sorte qu'il requière moins d'énergie pour se former, et font évoluer cet état de transition vers un complexe enzyme-produit (EP) qui se dissocie spontanément.
De plus, les enzymes peuvent coupler deux ou plusieurs réactions afin de permettre à une réaction thermodynamiquement défavorisée de se produire à l'aide d'une réaction thermodynamiquement favorisée de telle sorte que l'énergie combinée des produits de deux réactions soit inférieur à l'énergie combinée de leurs substrats. C'est très souvent le cas de l'hydrolyse de l'ATP, qui est généralement couplée à d'autres réactions chimiques, notamment du catabolisme (biosynthèses).
Cinétique enzymatique
La cinétique enzymatique étudie la façon dont les enzymes se lient à leurs substrats et les convertissent en produits de réaction. Les données quantitifant la cinétique d'une enzyme sont généralement obtenues à partir de dosages enzymatiques (en). En 1913, l'Allemand Leonor Michaelis et la Canadienne Maud Menten proposèrent une théorie quantitative de la cinétique enzymatique, appelée depuis équation de Michaelis-Menten[29]. Leur principale contribution a été de concevoir les réactions enzymatiques en deux étapes. Tout d'abord, les substrats se lient réversiblement à l'enzyme, formant un complexe enzyme-substrat. Puis l'enzyme catalyse la réaction chimique et libère les produits de réaction. Ces travaux ont ensuite été poursuivis par les Britanniques George Edward Briggs (en) et John B. S. Haldane (en), qui en dérivèrent les équations cinétiques encore largement utilisées de nos jours[30].


La vitesse d'une enzyme dépend des conditions de la solution et de la concentration en substrats. La vitesse maximum Vmax d'une réaction enzymatique peut être déterminé en augmentant la concentration [S] en substrats jusqu'à ce que la vitesse de formation des produits de réaction présente un plateau, comme représenté ci-contre. Cette saturation s'explique par le fait que plus la concentration en substrats augmente, plus ce substrat se lie aux enzymes, de sorte que la concentration en complexes enzyme-substrats augmente et la concentration en enzyme libre diminue ; la vitesse maximum de réaction correspond à la situation où toutes les enzymes sont liées à leurs substrats de sorte qu'il ne reste plus d'enzyme libre ayant des sites de liaison à occuper.
Outre la vitesse maximum Vmax d'une enzyme, la quantité de substrat nécessaire pour atteindre une vitesse de réaction donnée est une autre grandeur importante caractérisant l'activité d'une enzyme. Cette quantité est mesurée par la constante de Michaelis KM, qui représente la concentration de substrat nécessaire pour que l'enzyme atteigne la moitié de Vmax. Chaque enzyme possède généralement une Km donnée pour chacun de ses substrats. La vitesse v de réaction à concentration [S] de substrat, correspondant à l'augmentation de la concentration [P] en produit, est alors donnée par l'équation :
![v = \frac{d}{d t} [P] = \frac{ {[S]}}{K_\mathrm{M} + [S]} V_\max](../i/m/b8de9a893bbf26ddb85ffee4b27fe3c9.png) .
.
La constante catalytique, notée kcat, également appelée turnover number (TON), représente le nombre de molécules de substrat converties en produits par site actif et par seconde. Elle est liée à la vitesse maximum Vmax et à la concentration [E] de l'enzyme par l'équation Vmax = kcat [E].
L'activité enzymatique est mesurée en katals, unité SI définie par 1 kat = 1 mol⋅s-1, ou, plus fréquemment, en unités enzymatiques, définies par 1 U = 1 µmol·min-1 : ces grandeurs représentent la quantité d'enzyme nécessaire pour traiter une quantité unitaire de substrat par unité de temps, dans des conditions opératoires qui doivent être précisées avec la mesure. On peut en déduire l'activité spécifique de l'enzyme, qui représente son activité par unité de masse, exprimée par exemple en U⋅mg-1.
L'efficacité d'une enzyme peut être exprimée en termes de kcat / KM, qui représente la constante de spécificité. Dans la mesure où elle reflète à la fois l'affinité pour les substrats et l'efficacité de la catalyse, elle est utile pour comparer des enzymes entre elles ou pour comparer la même enzyme par rapport à différents substrats. Le maximum de la constante de spécificité est appelée limite de diffusion et se situe aux alentours de 108 à 109 M-1⋅s-1. À cette valeur, chaque contact entre l'enzyme et ses substrats conduit à une réaction chimique, et la vitesse de formation des produits de réaction n'est plus limitée par la vitesse de réaction mais par la vitesse de diffusion. Les enzymes qui présentent de telles propriétés sont appelées enzymes parfaites. Ce sont par exemple la triose-phosphate isomérase, l'anhydrase carbonique, l'acétylcholinestérase, la catalase, la fumarase, les β-lactamases, ou encore les superoxyde dismutases. Le turnover de telles enzymes peut atteindre plusieurs millions de réactions par seconde et par site actif.
La cinétique de Michaelis-Menten repose sur la loi d'action de masse, qui dérive de l'hypothèse que la diffusion de la matière est libre et que les collisions entre particules sont aléatoires et décrites par la thermodynamique. Cependant, de nombreux processus biochimiques ou cellulaires dérivent significativement de ces conditions en raison de la concentration très élevée des espèces chimiques dans le cytosol qui restreignent leur liberté de mouvement[31]. La cinétique de Michaelis-Menten a fait l'objet d'extensions récentes qui tentent de tenir compte de ces effets[32].
Inhibition enzymatique
Un inhibiteur enzymatique est une petite molécule qui réduit la vitesse de réaction d'une enzyme.
Types d'inhibition enzymatique
On range habituellement les types d'inhibition enzymatique dans les catégories suivantes.
Inhibition compétitive

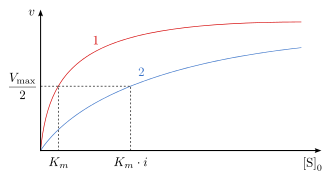
Un inhibiteur compétitif peut se lier à l'enzyme en empêchant ses substrats de le faire[33]. Il s'agit souvent d'une molécule qui ressemble à l'un des substrats et prend sa place sur l'un des sites de liaison mais sans que l'enzyme puisse catalyser la réaction chimique avec cet inhibiteur. Ainsi, le méthotrexate, un anticancéreux, est un inhibiteur compétitif de la dihydrofolate réductase, qui catalyse la réduction du dihydrofolate en tétrahydrofolate. Ce type d'inhibition peut être contourné par une concentration élevée en substrat. Il peut également s'agir d'une molécule qui se lie sur un autre site de l'enzyme et induit des changements conformationnels qui modifient les propriétés du site de liaison au substrat par effet allostérique. En conséquence, l'affinité de l'enzyme pour ses substrats diminue et sa constante de Michaelis KM augmente, tandis que sa vitesse maximum Vmax demeure inchangée.

Inhibition non compétitive

Un inhibiteur non compétitif se lie à l'enzyme sur un site indépendant des sites de liaison aux substrats. Ceux-ci se lient donc à l'enzyme avec une affinité inchangée, de sorte que la constante de Michaelis KM demeure inchangée. Cependant, l'inhibiteur réduit l'efficacité de l'enzyme, c'est-à-dire sa constante catalytique kcat, et, par conséquent, sa vitesse maximum Vmax. Contrairement à l'inhibition compétitive, l'inhibition non compétitive n'est pas réduite par l'augmentation de la concentration du substrat.
Inhibition incompétitive
Un inhibiteur incompétitif ne peut se lier qu'au complexe enzyme-substrat et non à l'enzyme seule. Ce type d'inhibiteurs est par conséquent d'autant plus efficace que la concentration en substrats est élevée. Le complexe enzyme-substrat-inhibiteur est inactif et ne peut catalyser la conversion des substrats en produits. Ce type d'inhibition demeure rare[34].
Inhibition mixte
Un inhibiteur mixte se lie à un site allostérique distinct du site de liaison des substrats sur l'enzyme, et ces deux liaisons interagissent l'une sur l'autre. La fonctionnalité de l'enzyme est réduite mais pas supprimée lorsqu'elle est liée à l'inhibiteur. Ce type d'inhibiteur ne suit pas l'équation de Michaelis-Menten.
Inhibition irréversible
Un inhibiteur irréversible, également appelé inhibiteur suicide, se lie à l'enzyme pour l'inhiber de façon permanente, généralement à travers une liaison covalente. La pénicilline[35] et l'aspirine[36] agissent de cette façon respectivement sur les transpeptidases et les cyclo-oxygénases.
Rôle biochimique
Chez de nombreux êtres vivants, les inhibiteurs enzymatiques interviennent dans le cadre d'un mécanisme général de rétroaction métabolique. Lorsqu'une molécule est produite en excès, elle peut agir comme inhibiteur de l'enzyme qui engage la voie métabolique produisant cette molécule, ce qui a pour effet d'en réduire la production et d'en maintenir la concentration physiologique à un niveau convenable. Il s'agit d'une forme de rétraction négative. Des voies métaboliques majeures telles que le cycle de Krebs utilisent des mécanismes de ce type.
Dans la mesure où les inhibiteurs modulent l'activité de certaines enzymes, ils sont fréquemment employés comme médicaments. De nombreux médicaments sont des inhibiteurs compétitifs réversibles qui ressemblent au substrat naturel de ces enzymes. Outre le méthotrexate présenté plus haut, de tels inhibiteurs compétitifs sont par exemple les statines utilisée pour traiter l'hypercholestérolémie[37] en inhibant l'HMG-CoA réductase et les inhibiteurs de protéase utilisés pour traiter les infections à rétrovirus tels que le VIH[38]. Un exemple classique d'inhibition irréversible est celui de l'aspirine, qui inhibe les cyclo-oxygénases COX-1 (en) et COX-2 (en) produisant les messagers de l'inflammation que sont les prostaglandines[36].
D'autres inhibiteurs enzymatiques sont des poisons. C'est par exemple le cas du cyanure CN–, qui se lie au cuivre et au fer du site actif de la cytochrome c oxydase[39] et bloque la respiration cellulaire.
Fonction biologique
Les enzymes remplissent un grand nombre de fonctions au sein des êtres vivants. Elles sont indispensables aux mécanismes de transduction de signal et de régulation des processus cellulaires, souvent à travers l'activité de kinases et de phosphatases[40]. Elles interviennent également dans la génération de mouvements, comme la myosine qui hydrolyse l'ATP lors de la contraction musculaire et permet le transport de molécules à travers la cellule en agissant sur le cytosquelette[41]. Les pompes à ions des membranes cellulaires sont d'autres ATPases qui interviennent dans le transport actif transmembranaire. Des enzymes interviennent également dans des processus plus exotiques tels que la bioluminescence produite par exemple par la luciférase chez les lucioles, ou encore par certaines bactéries[42]. Les virus contiennent quant à eux des enzymes leur permettant d'infecter des cellules, comme l'intégrase et la transcriptase inverse du VIH, ou de sortir des cellules infectées comme la neuraminidase du virus de la grippe[43].
Digestion
Les enzymes jouent un rôle important dans l'appareil digestif humain, où des enzymes telles que les amylases et les peptidases interviennent en dégradant des biopolymères comme l'amidon et les protéines en petites molécules susceptibles d'être absorbées au niveau des intestins — respectivement en maltose (puis en glucose) et en acides α-aminés dans notre exemple. Les macromolécules biologiques sont en effet trop grosses pour être absorbées directement, et ce sont leurs monomères qui sont absorbés. La digestion recouvre précisément ce processus de clivage des macromolécules en petites molécules. Des enzymes différentes sont nécessaires pour digérer des substances différentes. Chez les ruminants, qui sont herbivores, des microorganismes de l'intestin produisent une enzyme particulière, la cellulase, capable de cliver la cellulose de la paroi cellulaire des cellules végétales[44].
Métabolisme

Plusieurs enzymes peuvent travailler ensemble dans un ordre défini pour former des voies métaboliques : dans une telle configuration, un produit d'une enzyme devient un substrat de l'enzyme suivante. Il est possible que plusieurs enzymes catalysent parallèlement la même réaction ; ceci permet des modes de régulation plus complexes avec, par exemple, une faible constante d'activité pour une enzyme mais une seconde enzyme pouvant atteindre un niveau d'activité élevé lorsqu'elle est activée[45].
Les enzymes déterminent les étapes des voies métaboliques. En l'absence d'enzymes, le métabolisme n'emprunterait pas les mêmes chemins et ne pourrait pas être régulé afin d'être en cohérence avec les besoins de la cellule. La plupart des voies métaboliques principales du métabolisme sont régulées au niveau de quelques étapes clés, généralement au niveau d'enzymes qui requièrent l'hydrolyse de l'ATP. Cette réaction étant fortement exothermique (c'est-à-dire qu'elle s'accompagne d'une variation d'enthalpie libre élevée), elle peut être couplée à une réaction endothermique (c'est-à-dire s'accompagnant d'une variation d'enthalpie libre négative) afin de la rendre thermodynamiquement favorable.
Contrôle de l'activité enzymatique
Il existe essentiellement cinq moyens de contrôler l'activité des enzymes dans les cellules.
Régulation
Les enzymes peuvent être activées ou inhibées par d'autres molécules. Le ou les produits finaux d'une voie métabolique sont souvent des inhibiteurs de l'une des premières enzymes de cette voie, généralement la première enzyme qui catalyse une étape irréversible, ce qui régule la quantité de produit final ; il s'agit d'un mécanisme de rétroaction, dans la mesure où la quantité de produit final est régulée par la propre concentration de ce produit. La rétroaction permet d'ajuster efficacement le niveau de biosynthèse d'un ensemble de métabolites intermédiaires en fonction des besoins de la cellule, en évitant de produire un excès de molécules qui serait perdu et réduirait l'efficacité globale du métabolisme cellulaire.
Modification post-traductionnelle
La phosphorylation, la myristoylation et la glycosylation sont des exemples de modifications post-traductionnelles. Ainsi, la phosphorylation, induite par l'insuline, de nombreuses enzymes, dont la glycogène synthase, permet de contrôler l'anabolisme et le catabolisme du glycogène et permet à la cellule de s'adapter aux variations de la glycémie[46].
Le clivage d'une chaîne polypeptidique est un autre exemple de modification post-traductionnelle. La chymotrypsine, une peptidase digestive, est produite dans le pancréas sous une forme inactive appelée chymotrypsinogène (en) et transportée sous cette forme jusque dans l'estomac, où elle est activée. Ceci permet d'éviter que la chymotrypsine active ne digère d'autres tissus avant de parvenir dans l'estomac. Ce type de précurseur inactif d'une enzyme est un zymogène.
Quantité
La production des enzymes peut être accrue ou réduite par la cellule en réponse à des modifications dans son environnement. Cette forme de régulation de l'expression génétique est appelée induction enzymatique. C'est par exemple le cas des bactéries qui deviennent résistantes à des antibiotiques, par exemple à la pénicilline par induction d'enzymes appelées β-lactamases, lesquelles hydrolysent le noyau β-lactame[47] qui est précisément le pharmacophore de ce type d'antibiotiques. Les cytochrome P450 oxydases sont un autre exemple d'induction enzymatique. Ces enzymes jouent un rôle important dans la métabolisation de nombreux médicaments, et leur induction ou leur inhibition peuvent conduire à des interactions médicamenteuses.
La quantité d'enzymes présente dans une cellule peut également être modulée par leur dégradation.
Distribution subcellulaire
La distribution intracellulaire des enzymes peut être compartimentée, différentes voies métaboliques se déroulant dans différents compartiments cellulaires. Ainsi, les acides gras sont produits par un ensemble d'enzymes distribuées dans le cytosol, le réticulum endoplasmique et l'appareil de Golgi, et sont dégradés pour en libérer l'énergie chimique par β-oxydation sous l'effet d'un autre ensemble d'enzymes situées dans les mitochondries[48]. De plus, les différents compartiments d'une cellule connaissent des niveaux de protonation différents (par exemple, les lysosomes sont acides quand le cytoplasme est neutre) ou des niveaux d'oxydation différents (par exemple, le périplasme est plus oxydant que le cytoplasme), ce qui module également le niveau d'activité des enzymes qui s'y trouvent.
Spécialisation par organes
Chez les organismes multicellulaires, les cellules de différents organes ou de différents tissus présentent différents modes d'expression génétique et produisent par conséquent différentes variantes, appelés isoenzymes, d'un même ensemble d'enzymes, catalysant différentes réactions métaboliques. Ceci offre un mécanisme de régulation du métabolisme global de l'organisme. Ainsi, l'hexokinase, première enzyme de la glycolyse, possède une forme spécialisée, la glucokinase, exprimée dans le foie et dans le pancréas, dont l'affinité pour le glucose est plus faible mais qui est plus sensible aux variations de concentration de glucose[49]. Ceci permet à cette enzyme de réguler la production d'insuline en fonction des variations de la glycémie[50].
Pathologies
Dans la mesure où un contrôle des étroit de l'activité enzymatique est essentiel pour l'homéostasie de l'organisme, tout dysfonctionnement (mutation, surproduction, sousproduction ou absence) d'une seule enzyme critique peut provoquer une maladie génétique. La dysfonctionnement d'un seul type d'enzyme parmi les milliers du corps humain peut être mortel : c'est par exemple le cas d'une déficience en hexosaminidase, responsable de la maladie de Tay-Sachs[51].
La forme la plus courante de phénylcétonurie est un autre exemple de maladie résultant d'une déficience enzymatique. De nombreuses mutations n'affectant chaque fois qu'une seul résidu d'acide aminé de la phénylalanine hydroxylase, qui catalyse la première étape de la dégradation de la phénylalanine, conduit à l'accumulation de cet acide aminé et de produits apparentés. Certaines de ces mutations affectent le site actif, altérant directement la liaison des substrats et la catalyse, mais de nombreuses autres mutations touchent des résidus éloignés du site actif et réduisent l'activité enzymatique par altération du repliement de l'enzyme (structure tertiaire) ou affectant son oligomérisation (structure quaternaire)[52],[53]. Ceci peut conduire au handicap mental si la maladie n'est pas prise en charge. L'administration orale d'enzymes peut traiter certaines déficiences enzymatiques fonctionnelles telles que l'insuffisance pancréatique exocrine (en)[54] et l'intolérance au lactose[55].
Des maladies peuvent résulter d'autres types de dysfonctionnements enzymatiques lorsque ces derniers affectent les enzymes assurant la réparation de l'ADN, ce qui provoque des mutations dans les cellules germinales. De tels défauts enzymatiques sont davantage susceptibles de provoquer des cancers parce que les cellules deviennent alors plus sensibles aux mutations affectant leur génome. La lente accumulation de telles mutations peut alors conduire à l'apparition de cancers. Un exemple de tels syndromes cancéreux héréditaires est le xeroderma pigmentosum, qui conduit à l'apparition d'un cancer de la peau à la suite d'une exposition même minime à la lumière ultraviolette[56].
Utilisations industrielles
Certaines enzymes sont utilisées dans l'industrie chimique et pour d'autres applications industrielles lorsque des catalyseurs très spécifiques sont nécessaires. Les enzymes naturelles sont cependant assez limitées du point de vue des réactions qu'elles sont capables de catalyser, car il s'agit de réactions spécifiques au métabolisme des êtres vivants, et non de l'industrie chimique en général ; les enzymes sont également actives dans les conditions physico-chimiques physiologiques des organismes dont elles sont issus, conditions qui diffèrent souvent de celles mises en œuvre dans le cadre de procédés industriels. Par conséquent, le génie protéique (en) est un domaine de recherche actif qui vise à développer de nouvelles enzymes dotées de propriétés innovantes, que ce soit par conception rationnelle ou par évolution in vitro[57],[58]. Depuis le début du siècle, des enzymes ont ainsi pu être conçues de manière entièrement artificielle afin de catalyser des réactions chimiques qui ne se produisent pas naturellement[59].
Le tableau ci-dessous résume quelques applications industrielles de certaines enzymes courantes.
| Application industrielle | Enzymes employées | Utilisations |
|---|---|---|
| Industrie des biocarburants | Cellulases | Dégradation de la cellulose en glucides simples qui peuvent être fermentés pour produire de l'éthanol cellulosique[60]. |
| Ligninases | Prétraitement de la biomasse pour la production de biocarburants[60]. | |
| Lessive biologique (en) | Peptidases, amylases, lipases | Élimine les protéines, l'amidon, les taches de graisse ou d'huile de la vaisselle ou du linge[61]. |
| β-Mannosidases | Homogénéisation des préparations à base de gomme de guar[61]. | |
| Brassage | Amylase, glucanases, peptidases | Clivage des polysaccharides et des polypeptides du malt[62]:150–9. |
| β-Glucanases | Amélioration des propriétés de filtration du moût et de la bière[62]:545. | |
| Amylases et pullulanases | Production de bières basse calories et ajustement des caractéristiques de fermentation[62]:575. | |
| Acétolactate décarboxylase (ALDC) | Amélioration de l'efficacité de la fermentation en réduisant la formation de diacétyle[63]. | |
| Cuisson des aliments | Papaïne | Utilisation comme attendrisseur pour favoriser la tendreté de la viande en cuisine[64]. |
| Industrie laitière | Rennine | Hydrolyse des protéines lors de la production de fromages[65]. |
| Lipases | Production des camemberts et des bleus tels que le roquefort[66]. | |
| Processus agroalimentaires | Amylases | Production de sucres à partir d'amidon, par exemple pour produire du sirop de maïs à haute teneur en fructose[67]. |
| Peptidases | Réduction de la teneur en protéines de la farine, par exemple lors de la fabrication de biscuits[68]. | |
| Trypsine | Production de nourriture hypoallergénique (en) pour bébés[68]. | |
| Cellulases, pectinases | Amélioration de la clarté des jus de fruits[69]. | |
| Biologie moléculaire | Nucléases, ADN ligase et ADN polymérases | Utilisation d'enzymes de restriction et réaction en chaîne par polymérase afin de créer des ADN recombinants[70]:6.2. |
| Industrie papetière | Xylanases, hémicellulases et lignine peroxydases | Élimination de la lignine du papier kraft[71]. |
| Hygiène | Peptidases | Nettoyage des protéines des lentilles de contact afin de prévenir les infections[72]. |
| Traitement de l'amidon | Amylases | Conversion de l'amidon en glucose et divers sirops à sucre inverti[73]. |
Histoire et nomenclature
Découverte de la diastase

La première enzyme, la diastase, a été isolée en 1833 par Anselme Payen et Jean-François Persoz[74]. Après avoir traité un extrait aqueux de malt à l'éthanol, ils précipitèrent une substance sensible à la chaleur et capable d'hydrolyser l'amidon, d'où son nom de diastase forgé à partir du grec ancien ἡ διάστασις désignant l'action de cliver. Il s'agissait en réalité d'une amylase.
Le biologiste et chimiste Émile Duclaux (1840-1904) préconisa à la fin du XIXe siècle de nommer les substances actives semblables à la diastase à l'aide du suffixe -ase en référence à cette dernière[75].
Notions de ferments et de zymases


Quelques décennies plus tard, alors qu'il étudiait la fermentation alcoolique du sucre par une levure, Louis Pasteur conclut qu'un principe actif — qu'il appela ferment — contenu dans la levure était responsable de cette fermentation. Il considérait que cette substance n'était active qu'au sein d'une cellule vivante. Pasteur écrivit[76],[77] :
« la fermentation alcoolique est un acte en corrélation avec la vie et l'organisation des cellules de levure, et non avec la mort ou la putréfaction ; que ce n'est pas non plus un phénomène de contact, cas dans lequel la transformation du sucre s'accomplirait sans rien lui abandonner ni rien lui prendre. »
En 1877, le physiologiste allemand Wilhelm Kühne (en) (1837–1900) introduisit le terme enzyme en référence à ce processus, du grec ancien ἔνζυμον forgé à partir du préfixe ἐν « dans » et du substantif ἡ ζύμη « levain ». Le mot enzyme désigna par la suite les substances actives non vivantes telles que la pepsine, tandis que le mot ferment était utilisé en référence à l'activité chimique produites par des être vivants.
Le chimiste allemand Eduard Buchner publia son premier article sur l'étude des extraits de levure en 1897. Au cours d'une série d'expériences à l'université Humboldt de Berlin, il découvrit que le sucre pouvait être fermenté par des extraits de levure même en l'absence de toute cellule de levure dans le mélange, et reçut en 1907 le prix Nobel de chimie pour sa découverte de la fermentation sans cellule vivante[78]. Il appela zymase l'enzyme responsable de la fermentation, reprenant la construction du nom des enzymes en se référant au processus qu'elles catalysent auquel est adjoint le suffixe -ase, suivant en cela les recommandations d'Émile Duclaux quelques années plus tôt.
Caractérisation biochimique
La nature biochimique des enzymes demeurait cependant encore inconnue au début du XXe siècle. De nombreux scientifiques avaient observé que l'activité enzymatique était associée aux protéines, tandis que d'autres (dont Richard Willstätter, prix Nobel de chimie 1915 pour ses travaux sur la chlorophylle) considéraient que les protéines étaient de simples véhicules de l'activité enzymatique, étant par elles-mêmes incapables de catalyser des réactions chimiques[79],[80]. En 1926, James B. Sumner montra que l'uréase était une enzyme de nature purement protéique et la cristallisa ; il fit de même en 1937 avec la catalase. John Howard Northrop et Wendell Meredith Stanley achevèrent d'établir la nature protéique des enzymes en travaillant sur la pepsine (1930), la trypsine et la chymotrypsine. Ces trois chercheurs partagèrent le prix Nobel de chimie de 1946[81].
Le fait que des enzymes puissent être cristallisées permit d'établir leur structure tridimensionnelle par cristallographie aux rayons X. Ceci fut réalisé pour la première fois avec le lysozyme, une enzyme présente dans les larmes, la salive et le blanc d'œuf qui digère l'enveloppe de certaines bactéries : sa structure fut résolue par une équipe dirigée par David Chilton Phillips (en) et publiée en 1965[82]. L'établissement à haute résolution de la structure du lysozyme marqua les débuts de la biologie structurale et de l'étude du fonctionnement des enzymes à l'échelle atomique[83].
Dénomination et classement
Le comité des nomenclatures de l'Union internationale de biochimie et de biologie moléculaire (NC-IUBMB) a développé la nomenclature EC (pour Enzyme Commission), fondée sur le type de réactions chimiques catalysées[84]. Cette nomenclature est constituée de quatre nombres séparés par des points, associés à une dénomination systématique unique ; par exemple, l'α-amylase a pour numéro EC et pour dénomination systématique 4-α-D-glucane glucanohydrolase. La dénomination systématique est rarement employée : on lui préfère généralement des appellations d'usage, une enzyme pouvant avoir plusieurs appellations, certaines étant parfois ambiguës.
Les enzymes sont classées en six principaux groupes, en fonction du type de réaction qu'elles catalysent :
| Groupe | Code | Type de réaction |
|---|---|---|
| Oxydoréductases | EC 1 | Réaction d'oxydoréduction |
| Transférases | EC 2 | Transfert de groupes fonctionnels d'un substrat à un autre |
| Hydrolases | EC 3 | Hydrolyses |
| Lyases | EC 4 | Rupture de différentes liaisons chimiques par des moyens autres que l'hydrolyse ou l'oxydation |
| Isomérases | EC 5 | Isomérisations |
| Ligases ou synthétases | EC 6 | Formations de liaisons covalentes couplée à l'hydrolyse d'un nucléoside triphosphate (généralement l'ATP) |
Un numéro EC fait référence à une réaction chimique donnée, mais pas à une molécule donnée : un même numéro EC peut ainsi correspondre à plusieurs isoenzymes, c'est-à-dire à plusieurs protéines catalysant la même réaction chimique mais ayant des séquences peptidiques différentes — c'est par exemple le cas de toutes les ADN polymérases, qui partagent toutes le numéro EC — tandis qu'une même enzyme peut avoir plusieurs numéros EC lorsque la protéine qui la constitue porte plusieurs sites actifs catalysant des réactions chimiques différentes — on parle par exemple d'enzyme bifonctionnelle lorsqu'une même protéine catalyse deux réactions chimiques, comme l'uridine monophosphate synthétase (UMPS) qui porte une sous-unité orotate phosphoribosyltransférase (EC ) et une sous-unité orotidine-5'-phosphate décarboxylase (EC ).
Le nom des enzymes fait le plus souvent référence à un ou plusieurs de ses substrats, parfois au type de réaction chimique catalysée, et très souvent avec le suffixe -ase, parfois avec le suffixe -ine. Les noms des enzymes sont dans ce cas du genre féminin ; le lysozyme et les ribozymes font exceptions, bien que le suffixe -zyme provienne du grec ancien ἡ ζύμη (« levain »), qui était du genre féminin. Le nom enzyme lui-même est employé au féminin[85], ainsi que ses composés (par exemple coenzyme[86]), bien que l'usage hésitât encore au siècle dernier. La lactase, l'alcool déshydrogénase, l'ADN polymérase, la triose-phosphate isomérase, la papaïne, la pepsine et la trypsine sont des exemples de noms d'enzymes. La glucose oxydase est ainsi une enzyme catalysant l'oxydation du glucose, tandis que l'amidon synthase catalyse la biosynthèse de l'amidon. Plusieurs enzymes qui catalysent la même réaction chimique sont appelées isoenzymes.
Terminologie
- Enzyme de la transformation alimentaire : enzyme employé pour contrôler la texture, le goût, l’aspect ou la valeur nutritive des aliments. Les amylases dégradent les complexes polysaccharidiques en sucres plus simples ; et les protéases « attendrissent » les protéines de la viande. Un objectif important de la biotechnologie alimentaire est le développement de nouvelles enzymes alimentaires améliorant la qualité de la transformation des aliments.
- Enzyme de restriction ou endonucléase de restriction : classe d’enzymes qui coupent l'ADN après avoir reconnu une séquence spécifique. Les trois types d’endonucléase de restriction sont :
- enzyme répressible : enzyme dont l’activité peut être réduite par la présence d’une molécule régulatrice ;
- enzyme limitante : enzyme dont l’activité contrôle le rendement du produit final d’une voie métabolique multi-enzymatique... ;
- enzyme constitutive : enzyme dont la concentration dans la cellule est constante et n'est pas influencée par une concentration en substrat.
Notes et références
- ↑ (en) Jacinta A. Lodge, Timm Maier, Wolfgang Liebl, Volker Hoffmann et Norbert Sträter, « Crystal Structure of Thermotoga maritima α-Glucosidase AglA Defines a New Clan of NAD+-dependent Glycosidases », Journal of Biological Chemistry, vol. 278, no 21, , p. 19151-19158 (PMID 12588867, DOI 10.1074/jbc.M211626200, lire en ligne)
- ↑ (en) Ida Schomburg, Antje Chang, Sandra Placzek, Carola Söhngen, Michael Rother, Maren Lang, Cornelia Munaretto, Susanne Ulas, Michael Stelzer, Andreas Grote, Maurice Scheer et Dietmar Schomburg, « BRENDA in 2013: integrated reactions, kinetic data, enzyme function data, improved disease classification: new options and contents in BRENDA », Nucleic Acids Research, vol. 41, no D1, , D764-D772 (PMID 23203881, DOI 10.1093/nar/gks1049, lire en ligne)
- ↑ (en) A. Radzicka et R. Wolfenden, « A proficient enzyme », Science, vol. 267, no 5194, , p. 90-93 (PMID 7809611, DOI 10.1126/science.7809611, lire en ligne)
- ↑ (en) Brian P. Callahan et Brian G. Miller, « OMP decarboxylase—An enigma persists », Bioorganic Chemistry, vol. 35, no 6, , p. 465-469 (PMID 17889251, DOI 10.1016/j.bioorg.2007.07.004, lire en ligne)
- ↑ (en) Christian B. Anfinsen, « Principles that govern the folding of protein chains », Science, vol. 181, no 4096, , p. 223-230 (PMID 4124164, DOI 10.1126/science.181.4096.223, lire en ligne)
- ↑ (en) Debra Dunaway-Mariano, « Enzyme Function Discovery », Structure, vol. 16, no 11, , p. 1599-1600 (PMID 19000810, DOI 10.1016/j.str.2008.10.001, lire en ligne)
- ↑ (en) L H Chen, G L Kenyon, F Curtin, S Harayama, M E Bembenek, G Hajipour et C P Whitman, « 4-Oxalocrotonate tautomerase, an enzyme composed of 62 amino acid residues per monomer », Journal of Biological Chemistry, vol. 267, no 25, , p. 17716-17721 (PMID 1339435, lire en ligne)
- ↑ (en) S. Smith, « The animal fatty acid synthase: one gene, one polypeptide, seven enzymes », The FASEB Journal, vol. 8, no 15, , p. 1248-1259 (PMID 8001737, lire en ligne)
- ↑ (en) Karl-Erich Jaeger et Thorsten Eggert, « Enantioselective biocatalysis optimized by directed evolution », Current Opinion in Biotechnology, vol. 15, no 4, , p. 305-313 (PMID 15358000, DOI 10.1016/j.copbio.2004.06.007, lire en ligne)
- ↑ (en) Igor V. Shevelev et Ulrich Hübscher, « The 3′–5′ exonucleases », Nature Reviews Molecular Cell Biology, vol. 3, no 5, , p. 364-376 (PMID 11988770, DOI 10.1038/nrm804, lire en ligne)
- ↑ (en) Nikolay Zenkin, Yulia Yuzenkova et Konstantin Severinov1, « Transcript-Assisted Transcriptional Proofreading », Science, vol. 313, no 5786, , p. 518-520 (PMID 16873663, DOI 10.1126/science.1127422, lire en ligne)
- ↑ (en) Michael Ibba et Dieter Söll, « Aminoacyl-tRNA synthesis », Annual Reviews Biochemistry, vol. 69, , p. 617-650 (PMID 10966471, DOI 10.1146/annurev.biochem.69.1.617, lire en ligne)
- ↑ (en) Marina V. Rodnina et Wolfgang Wintermeyer, « Fidelity of aminoacyl-tRNA selection on the ribosome: kinetic and structural mechanisms », Annual Reviews Biochemistry, vol. 70, , p. 415-435 (PMID 11395413, DOI 10.1146/annurev.biochem.70.1.415, lire en ligne)
- ↑ (en) Olga Khersonsky et Dan S. Tawfik, « Enzyme Promiscuity: A Mechanistic and Evolutionary Perspective », Annual Reviews Biochemistry, vol. 79, , p. 471-505 (PMID 20235827, DOI 10.1146/annurev-biochem-030409-143718, lire en ligne)
- ↑ (en) Patrick J O'Brien, Daniel Herschlag, « Catalytic promiscuity and the evolution of new enzymatic activities », Chemistry & Biology, vol. 6, no 4, , R91-R105 (PMID 10099128, DOI 10.1016/S1074-5521(99)80033-7, lire en ligne)
- ↑ (de) Emil Fischer, « Einfluss der Configuration auf die Wirkung der Enzyme », Berichte der deutschen chemischen Gesellschaft, vol. 27, no 3, , p. 2985-2993 (DOI 10.1002/cber.18940270364, lire en ligne)
- ↑ (en) Daniel E. Koshland, « Application of a Theory of Enzyme Specificity to Protein Synthesis », Proceedings of the National Academy of Sciences of the United States of America, vol. 44, no 2, , p. 98-104 (PMID 16590179, PMCID 335371, lire en ligne)
- ↑ (en) Andrea Vasella, , Gideon J Davies et Matthias Böhm, « Glycosidase mechanisms », Current Opinion in Chemical Biology, vol. 6, no 5, , p. 619-629 (DOI 10.1016/S1367-5931(02)00380-0, lire en ligne)
- ↑ (en) Arieh Warshel, Pankaz K. Sharma, Mitsunori Kato, Yun Xiang, Hanbin Liu et Mats H. M. Olsson, « Electrostatic Basis for Enzyme Catalysis », Chemical Reviews, vol. 106, no 8, , p. 3210-3235 (PMID 16895325, DOI 10.1021/cr0503106, lire en ligne)
- ↑ (en) Stephen J. Benkovic et Sharon Hammes-Schiffer, « A Perspective on Enzyme Catalysis », Science, vol. 301, no 5637, , p. 1196-1202 (PMID 12947189, DOI 10.1126/science.1085515, lire en ligne)
- ↑ (en) J. Villà, M. Štrajbl, T. M. Glennon, Y. Y. Sham, Z. T. Chu et A. Warshel, « How important are entropic contributions to enzyme catalysis? », Proceedings of the National Academy of Sciences of the United States of America, vol. 97, no 22, , p. 11899-11904 (PMID 11050223, PMCID 17266, DOI 10.1073/pnas.97.22.11899, lire en ligne)
- ↑ (en) Arvind Ramanathan, Andrej Savol, Virginia Burger, Chakra S. Chennubhotla et Pratul K. Agarwal, « Protein Conformational Populations and Functionally Relevant Substates », Accounts of Chemical Research, vol. 47, no 1, , p. 149-156 (PMID 23988159, DOI 10.1021/ar400084s, lire en ligne)
- ↑ (en) Chung-Jung Tsai, Antonio del Sol et Ruth Nussinov, « Protein allostery, signal transmission and dynamics: a classification scheme of allosteric mechanisms », Molecular BioSystems, vol. 5, no 3, , p. 207-216 (PMID 19225609, PMCID 2898650, DOI 10.1039/B819720B, lire en ligne)
- ↑ (en) Jean-Pierre Changeux et Stuart J. Edelstein, « Allosteric Mechanisms of Signal Transduction », Science, vol. 308, no 5727, , p. 1424-1428 (PMID 15933191, DOI 10.1126/science.1108595, lire en ligne)
- ↑ (en) Stefan Lüdtke, Piotr Neumann, Karl M. Erixon, Finian Leeper, Ronald Kluger, Ralf Ficner et Kai Tittmann, « Sub-ångström-resolution crystallography reveals physical distortions that enhance reactivity of a covalent enzymatic intermediate », Nature Chemistry, vol. 5, no 9, , p. 762-767 (PMID 23965678, DOI 10.1038/nchem.1728, lire en ligne)
- ↑ (en) Anne Chapman-Smith et John E. Cronan Jr, « The enzymatic biotinylation of proteins: a post-translational modification of exceptional specificity », Trends in Biochemical Sciences, vol. 24, no 9, , p. 359-363 (PMID 10470036, DOI 10.1016/S0968-0004(99)01438-3, lire en ligne)
- ↑ (en) Zoë Fisher, Jose A. Hernandez Prada, Chingkuang Tu, David Duda, Craig Yoshioka, Haiqian An, Lakshmanan Govindasamy, David N. Silverman et Robert McKenna, « Structural and Kinetic Characterization of Active-Site Histidine as a Proton Shuttle in Catalysis by Human Carbonic Anhydrase II », Biochemistry, vol. 44, no 4, , p. 1097-1105 (PMID 15667203, DOI 10.1021/bi0480279, lire en ligne)
- ↑ (en) Susanna Törnroth-Horsefield et Richard Neutze, « Opening and closing the metabolite gate », Proceedings of the National Academy of Sciences of the United States of America, vol. 105, no 50, , p. 19565-19566 (PMID 19073922, PMCID 2604989, DOI 10.1073/pnas.0810654106, lire en ligne)
- ↑ (en) Kenneth A. Johnson et Roger S. Goody, « The original Michaelis constant: translation of the 1913 Michaelis-Menten paper », Biochemistry, vol. 50, no 39, , p. 8264-8269 (PMID 21888353, PMCID 3381512, DOI 10.1021/bi201284u, lire en ligne)
- ↑ (en) George E. Briggs et John B. S. Haldane, « A Note on the Kinetics of Enzyme Action », Biochemical Journal, vol. 19, no 2, , p. 338-339 (PMID 16743508, PMCID 1259181)
- ↑ (en) R.John Ellis, « Macromolecular crowding: obvious but underappreciated », Trends in Biochemical Sciences, vol. 26, no 10, octobre, p. 2001 (PMID 11590012, DOI 10.1016/S0968-0004(01)01938-7, lire en ligne)
- ↑ (en) Raoul Kopelman, « Fractal Reaction Kinetics », Science, vol. 241, no 4873, , p. 1620-1626 (PMID 17820893, DOI 10.1126/science.241.4873.1620, lire en ligne)
- ↑ (en) Nicholas C. Price, « What is meant by ‘competitive inhibition’? », Trends in Biochemical Sciences, vol. 4, no 11, , N272-N273 (DOI 10.1016/0968-0004(79)90205-6, lire en ligne)
- ↑ (en) Athel Cornish-Bowden, « Why is uncompetitive inhibition so rare? », FEBS Letters, vol. 203, no 1, , p. 3-6 (PMID 3720956, DOI 10.1016/0014-5793(86)81424-7, lire en ligne)
- ↑ (en) J. F. Fisher, S. O. Meroueh et S. Mobashery, « Bacterial resistance to beta-lactam antibiotics: compelling opportunism, compelling opportunity », Chemical Reviews, vol. 105, no 2, , p. 395-424 (PMID 15700950, DOI 10.1021/cr030102i, lire en ligne)
- 1 2 (en) Douglas S Johnson, Eranthie Weerapana et Benjamin F Cravatt, « Strategies for discovering and derisking covalent, irreversible enzyme inhibitors », Future Medicinal Chemistry, vol. 2, no 6, , p. 949-964 (PMID 20640225, DOI 10.4155/fmc.10.21, lire en ligne)
- ↑ (en) Akira Endo, « The discovery and development of HMG-CoA reductase inhibitors », Journal of Lipid Research, vol. 33, no 11, , p. 1569-1582 (PMID 1464741, lire en ligne)
- ↑ (en) Alexander Wlodawer et Jiri Vondrasek, « INHIBITORS OF HIV-1 PROTEASE: A Major Success of Structure-Assisted Drug Design1 », Annual Review of Biophysics and Biomolecular Structure, vol. 27, , p. 249-284 (PMID 9646869, DOI 10.1146/annurev.biophys.27.1.249, lire en ligne)
- ↑ (en) S. Yoshikawa et W. S. Caughey, « Infrared evidence of cyanide binding to iron and copper sites in bovine heart cytochrome c oxidase. Implications regarding oxygen reduction », Journal of Biological Chemistry, vol. 265, no 14, , p. 7945-7958 (PMID 2159465, lire en ligne)
- ↑ (en) Tony Hunter, « Protein kinases and phosphatases: The Yin and Yang of protein phosphorylation and signaling », Cell, vol. 80, no 2, , p. 225-236 (PMID 7834742, DOI 10.1016/0092-8674(95)90405-0, lire en ligne)
- ↑ (en) Jonathan S. Berg, Bradford C. Powell et Richard E. Cheney, « A Millennial Myosin Census », Molecular Biology of the Cell, vol. 12, no 4, , p. 780-794 (PMID 11294886, PMCID 32266, DOI 10.1091/mbc.12.4.780, lire en ligne)
- ↑ (en) E. A. Meighen, « Molecular biology of bacterial bioluminescence », Microbiology and Molecular Biology Reviews, vol. 55, no 1, , p. 123-142 (PMID 2030669, PMCID 372803, lire en ligne)
- ↑ (en) E. de Clerq, « Highlights in the Development of New Antiviral Agents », Mini Reviews in Medicinal Chemistry, vol. 2, no 2, , p. 163-175 (PMID 12370077, DOI 10.2174/1389557024605474)
- ↑ (en) Roderick I. Mackie et Bryan A. White, « Recent Advances in Rumen Microbial Ecology and Metabolism: Potential Impact on Nutrient Output », Journal of Dairy Science, vol. 73, no 10, , p. 2971-2995 (PMID 2178174, DOI 10.3168/jds.S0022-0302(90)78986-2, lire en ligne)
- ↑ (en) Carol A. Rouzer et Lawrence J. Marnett, « Cyclooxygenases: structural and functional insights », Journal of Lipid Research, vol. 50, , S29-S34 (PMID 18952571, PMCID 2674713, DOI 10.1194/jlr.R800042-JLR200, lire en ligne)
- ↑ (en) Bradley W. Doble et James R. Woodgett, « GSK-3: tricks of the trade for a multi-tasking kinase », Journal of Cell Science, vol. 116, no Part 7, , p. 1175-1186 (PMID 12615961, PMCID 3006448, DOI 10.1242/jcs.00384, lire en ligne)
- ↑ (en) Peter M. Bennett et Ian Chopra, « Molecular basis of beta-lactamase induction in bacteria », Antimicrobial Agents and Chemotherapy, vol. 37, no 2, , p. 153-158 (PMID 8452343, PMCID 187630, DOI 10.1128/AAC.37.2.153, lire en ligne)
- ↑ (en) Nils Joakim Færgeman et Jens Knudsen, « Role of long-chain fatty acyl-CoA esters in the regulation of metabolism and in cell signalling », Biochemical Journal, vol. 33, no Part 1, , p. 1-12 (PMID 9173866, PMCID 1218279)
- ↑ (en) Kenji Kamata, Morihiro Mitsuya, Teruyuki Nishimura, Jun-ichi Eiki et Yasufumi Nagata, « Structural Basis for Allosteric Regulation of the Monomeric Allosteric Enzyme Human Glucokinase », Structure, vol. 12, no 3, , p. 429-438 (PMID 15016359, DOI 10.1016/j.str.2004.02.005, lire en ligne)
- ↑ (en) Philippe Froguel, Habib Zouali, Nathalie Vionnet, Gilberto Velho, Martine Vaxillaire, Fang Sun, Suzanne Lesage, Markus Stoffel, Jun Takeda, Philippe Passa, M. Alan Permutt, Jacques S. Beckmann, Graeme I. Bell et Daniel Cohen, « Familial Hyperglycemia Due to Mutations in Glucokinase — Definition of a Subtype of Diabetes Mellitus », The New England Journal of Medicine, vol. 328, no 10, , p. 697-702 (PMID 8433729, DOI 10.1056/NEJM199303113281005, lire en ligne)
- ↑ (en) Shintaro Okada et John S. O'Brien, « Tay-Sachs Disease: Generalized Absence of a Beta-D-N-Acetylhexosaminidase Component », Science, vol. 165, no 3894, , p. 698-700 (PMID 5793973, DOI 10.1126/science.165.3894.698, lire en ligne)
- ↑ (en) Heidi Erlandsen et Raymond C. Stevens, « The Structural Basis of Phenylketonuria », Molecular Genetics and Metabolism, vol. 68, no 2, , p. 103-125 (PMID 10527663, DOI 10.1006/mgme.1999.2922, lire en ligne)
- ↑ (en) T. Flatmark et R. C. Stevens, « Structural Insight into the Aromatic Amino Acid Hydroxylases and Their Disease-Related Mutant Forms », Chemical Reviews, vol. 99, no 8, , p. 2137-2160 (PMID 11849022, DOI 10.1021/cr980450y, lire en ligne)
- ↑ (en) Aaron Fieker, Jessica Philpott et Martine Armand, « Enzyme replacement therapy for pancreatic insufficiency: present and future », Clinical and Experimental Gastroenterology, vol. 4, , p. 55-73 (PMID 21753892, DOI 10.2147/CEG.S17634, lire en ligne)
- ↑ (en) Benjamin Misselwitz, Daniel Pohl, Heiko Frühauf, Michael Fried, Stephan R. Vavricka et Mark Fox, « Lactose malabsorption and intolerance: pathogenesis, diagnosis and treatment », United European Gastroenterology Journal, vol. 1, no 3, , p. 151-159 (PMID 24917953, PMCID 4040760, DOI 10.1177/2050640613484463, lire en ligne)
- ↑ (en) J. E. Cleaver, « Defective Repair Replication of DNA in Xeroderma Pigmentosum », Nature, vol. 218, no 5142, , p. 652-656 (PMID 5655953, DOI 10.1038/218652a0, lire en ligne)
- ↑ (en) V. Renugopalakrishnan, R. Garduño-Juárez, G. Narasimhan, C. S. Verma, X. Wei et Pingzuo Li, « Rational Design of Thermally Stable Proteins: Relevance to Bionanotechnology », Journal of Nanoscience and Nanotechnology, vol. 5, no 11, , p. 1759-1767 (PMID 16433409, DOI 10.1166/jnn.2005.441, lire en ligne)
- ↑ (en) Karl Hult et Per Berglund, « Engineered enzymes for improved organic synthesis », Current Opinion in Biotechnology, vol. 14, no 4, , p. 395-400 (PMID 12943848, DOI 10.1016/S0958-1669(03)00095-8, lire en ligne)
- ↑ (en) Lin Jiang, Eric A. Althoff, Fernando R. Clemente, Lindsey Doyle, Daniela Röthlisberger, Alexandre Zanghellini, Jasmine L. Gallaher, Jamie L. Betker, Fujie Tanaka, Carlos F. Barbas III, Donald Hilvert, Kendall N. Houk, Barry L. Stoddard et David Baker, « De novo computational design of retro-aldol enzymes », Science, vol. 319, no 5868, , p. 1387-1391 (PMID 18323453, PMCID 3431203, DOI 10.1126/science.1152692, lire en ligne)
- 1 2 (en) Sun Y, Cheng J, « Hydrolysis of lignocellulosic materials for ethanol production: a review », Bioresource Technology, vol. 83, no 1, , p. 1–11 (PMID 12058826, DOI 10.1016/S0960-8524(01)00212-7)
- 1 2 (en) Kirk O, Borchert TV, Fuglsang CC, « Industrial enzyme applications », Current Opinion in Biotechnology, vol. 13, no 4, , p. 345–351 (DOI 10.1016/S0958-1669(02)00328-2)
- 1 2 3 Dennis E. Briggs, Malts and Malting, London, Blackie Academic, , 1st éd. (ISBN 978-0412298004).
- ↑ (en) Dulieu C, Moll M, Boudrant J, Poncelet D, « Improved performances and control of beer fermentation using encapsulated alpha-acetolactate decarboxylase and modeling », Biotechnology Progress, vol. 16, no 6, , p. 958–65 (PMID 11101321, DOI 10.1021/bp000128k)
- ↑ Rodrigo Tarté, Ingredients in Meat Products Properties, Functionality and Applications, New York, Springer, (ISBN 978-0-387-71327-4), p. 177.
- ↑ (en) « Chymosin - GMO Database », GMO Compass, European Union, (consulté le 1er mars 2015)
- ↑ (en) Molimard P, Spinnler HE, « Review: Compounds Involved in the Flavor of Surface Mold-Ripened Cheeses: Origins and Properties », Journal of Dairy Science, vol. 79, no 2, feb 1996, p. 169–184 (DOI 10.3168/jds.S0022-0302(96)76348-8)
- ↑ (en) Guzmán-Maldonado H, Paredes-López O, « Amylolytic enzymes and products derived from starch: a review », Critical Reviews in Food Science and Nutrition, vol. 35, no 5, , p. 373–403 (PMID 8573280, DOI 10.1080/10408399509527706)
- 1 2 (en) « Protease - GMO Database », GMO Compass, European Union, (consulté le 28 février 2015)
- ↑ (en) Alkorta I, Garbisu C, Llama MJ, Serra JL, « Industrial applications of pectic enzymes: a review », Process Biochemistry, vol. 33, no 1, , p. 21–28 (DOI 10.1016/S0032-9592(97)00046-0)
- ↑ Stryer L, Berg JM, Tymoczko JL, Biochemistry, San Francisco, W.H. Freeman, , 5th éd. (ISBN 0-7167-4955-6, lire en ligne).
- ↑ (en) Bajpai P, « Application of enzymes in the pulp and paper industry », Biotechnology Progress, vol. 15, no 2, , p. 147–157 (PMID 10194388, DOI 10.1021/bp990013k)
- ↑ (en) Begley CG, Paragina S, Sporn A, « An analysis of contact lens enzyme cleaners », Journal of the American Optometric Association, vol. 61, no 3, , p. 190-4 (PMID 2186082)
- ↑ Paul L. Farris, Roy L. Whistler (dir.) et James N. BeMiller (dir.), Starch Chemistry and Technology, London, Academic, , 3rd éd. (ISBN 9780080926551), « Economic Growth and Organization of the U.S. Starch Industry ».
- ↑ Payen et Persoz, « Mémoire sur la diastase, les principaux produits de ses réactions et leurs applications aux arts industriels », Annales de chimie et de physique, 2e série, t. 53, 1833, p. 73-92, consultable sur Google Books.
- ↑ Traité de Microbiologie, vol. 2, éditions Masson, 1899, Paris ; chapitre 1, page 9 notamment.
- ↑ François Gros, « Biochimie cellulaire : Leçon inaugurale prononcée le mardi 15 janvier 1974 », sur Publications du Collège de France, OpenEdition, (consulté le 28 mai 2015), p. 4-33
- ↑ (en) Keith L. Manchester, « Louis Pasteur (1822–1895) — chance and the prepared mind », Trends in Biotechnology, vol. 13, no 12, , p. 511-515 (PMID 8595136, DOI 10.1016/S0167-7799(00)89014-9, lire en ligne)
- ↑ (en) « The Nobel Prize in Chemistry 1907 : Eduard Buchner - Biographical », sur Nobel Prizes and Laureates (consulté le 28 mai 2015)
- ↑ (en) Richard Willstätter, « Faraday lecture. Problems and methods in enzyme research », Journal of the Chemical Society (Resumed), , p. 1359-1381 (DOI 10.1039/JR9270001359, lire en ligne)
- ↑ (en) David Blow, « So do we understand how enzymes work? », Structure, vol. 8, no 4, , R77-R81 (PMID 10801479, DOI 10.1016/S0969-2126(00)00125-8, lire en ligne)
- ↑ (en) « The Nobel Prize in Chemistry 1946 », sur Nobel Prizes and Laureates (consulté le 28 mai 2015)
- ↑ (en) C. C. F. Blake, D. F. Koenig, G. A. Mair, A. C. T. North, D. C. Phillips et V. R. Sarma, « Structure of Hen Egg-White Lysozyme: A Three-dimensional Fourier Synthesis at 2 Å Resolution », Nature, vol. 206, no 4986, , p. 757-761 (PMID 5891407, DOI 10.1038/206757a0, lire en ligne)
- ↑ (en) Louise N. Johnson et Gregory A. Petsko, « David Phillips and the origin of structural enzymology », Trends in Biochemical Sciences, vol. 24, no 7, , p. 287-289 (PMID 10390620, DOI 10.1016/S0968-0004(99)01423-1, lire en ligne)
- ↑ (en) Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB), in consultation with the IUPAC-IUBMB Joint Commission on Biochemical Nomenclature (JCBN), « Web Version of Enzyme Nomenclature : Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the Nomenclature and Classification of Enzymes by the Reactions they Catalyse », sur IUBMB, (consulté le 28 mai 2015)
- ↑ « enzyme », sur Larousse en ligne (consulté le 28 mai 2015)
- ↑ « coenzyme », sur Larousse en ligne (consulté le 28 mai 2015)
Voir aussi
Articles connexes
- Exoenzyme
- Endoenzyme
- Isoenzyme
- Enzyme artificielle
- Fermentation
- Œnologie
- Réaction enzymatique
Liens externes
- (en) BRENDA : base de données sur les enzymes
 Portail de la biochimie
Portail de la biochimie  Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire