Mitochondrie
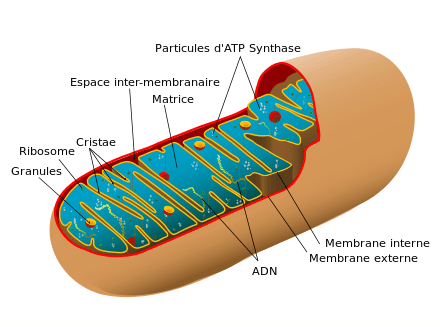
- ATP synthase ;
- espace intermembranaire mitochondrial ;
- matrice mitochondriale ;
- crêtes (cristae) ;
- ribosomes ;
- membrane mitochondriale interne ;
- membrane mitochondriale externe ;
- ADN mitochondrial.
Les mitochondries sont des organites présents dans la plupart des cellules d'eucaryotes[1]. Le terme mitochondrie provient du grec ancien μίτος (mitos) « fil » et χονδρίον (chondrion) « granule ». Leur taille varie généralement entre 0,5 et 1 μm de diamètre. Elles présentent des formes et des structures extrêmement variées. Elles sont invisibles lorsqu'elles ne sont pas teintes spécifiquement pour être observées. Elles sont souvent décrites comme les « centrales énergétiques » des cellules, dans la mesure où elles contribuent à l'essentiel de la production d'ATP cellulaire à travers la β-oxydation, le cycle de Krebs et la phosphorylation oxydative, l'ATP étant la molécule énergétique ubiquitaire utilisée dans un très grand nombre de réactions chimiques du métabolisme, et notamment de l'anabolisme.
Outre leur rôle dans le métabolisme énergétique cellulaire, les mitochondries interviennent également dans la signalisation cellulaire, la différenciation cellulaire et la mort cellulaire, ainsi que dans le contrôle du cycle cellulaire et de la croissance de la cellule[2]. Elles ont par ailleurs été associées à plusieurs maladies humaines, comme des maladies mitochondriales[3] et diverses cardiopathies[4].
Plusieurs propriétés des mitochondries en font des organites particuliers. Leur nombre par cellule varie considérablement par espèce, par tissu et par type cellulaire. Ainsi, les globules rouges du sang (hématies) sont totalement dépourvus de mitochondries, tandis que les cellules du foie peuvent en contenir plus de 2000. Cet organite est composé de plusieurs compartiments spécialisés dans plusieurs fonctions physiologiques : la membrane mitochondriale externe, l'espace intermembranaire mitochondrial, la membrane mitochondriale interne, et la matrice mitochondriale. Les protéines mitochondriales dépendent des espèces et des tissus considérés. Chez l'homme, les mitochondries cardiaques contiennent au moins 615 types de protéines différents[5], tandis qu'on en a identifié 940 chez le rat[6] ; le protéome mitochondrial est régulé de façon vraisemblablement dynamique[7].
Enfin, les mitochondries possèdent leur propre génome, dit génome mitochondrial, dont l'ADN présente de nombreuses analogies avec le génome des bactéries[8].
{kind=link}

Structure

1 : membrane interne,
2 : membrane externe,
3 : espace intermembranaire,
4 : matrice.
Les mitochondries ont une dimension de 1-2 à 10 μm de long et de 0,5 à 1 μm de diamètre. Elles se composent de deux membranes, une membrane mitochondriale externe et une membrane mitochondriale interne, qui délimitent trois milieux : le milieu extra-mitochondrial (cytoplasme de la cellule), l'espace intermembranaire mitochondrial, et la matrice mitochondriale.
Membrane externe
La membrane mitochondriale externe contient l'ensemble de l'organite et a une épaisseur d'environ 6 à 7,5 nm. Son rapport massique protéines/phospholipides est semblable à celui des membranes plasmiques des cellules d'eucaryotes, généralement voisin de 1:1. Elle contient un grand nombre de protéines membranaires intégrales (en) appelées porines et formant des canaux aqueux permettant aux molécules hydrophiles de moins de 5 kDa de diffuser librement à travers la bicouche lipidique[9] : anions, cations, acides gras, nucléotides peuvent ainsi traverser ce canal. Des protéines, plus massives, peuvent pénétrer dans la mitochondrie lorsqu'une séquence signal est attachée à leur extrémité N-terminale, permettant à ces protéines de se lier à une translocase de la membrane externe, laquelle assure leur transport actif à travers cette membrane[10].
La membrane externe contient également des enzymes impliquées dans des activités aussi diverses que la biosynthèse des acides gras (entrant notamment dans la constitution de la plupart des lipides), l'oxydation de l'adrénaline (une hormone et un neurotransmetteur) et la dégradation du tryptophane (un acide aminé protéinogène). Il s'agit notamment de la monoamine oxydase, de la NADH-cytochrome c réductase insensible à la roténone, de la kynurénine 7,8-hydroxylase et de l'acyl-CoA synthétase. La rupture de la membrane externe permet aux protéines de l'espace intermembranaire mitochondrial de se répandre dans le cytosol, conduisant à la mort cellulaire[11].
La membrane mitochondriale externe peut s'associer à la membrane du réticulum endoplasmique en une structure désignée par l'abréviation MAM (mitochondria-associated ER-membrane). Cette structure joue un rôle important dans certaines voies de signalisation cellulaire du calcium et intervient dans le transfert de lipides entre le réticulum endoplasmique et les mitochondries[12].
Espace intermembranaire
L'espace intermembranaire mitochondrial, parfois appelé espace périmitochondrial, est délimité par les membranes mitochondriales externes et internes. Dans la mesure où la membrane externe est perméable aux petites molécules, la concentration d'espèces chimiques telles que les oses et les ions (y compris les ions moléculaires de petite taille) est essentiellement la même dans l'espace intermembranaire que dans le cytosol. Les protéines doivent cependant porter une séquence spécifique de signalisation pour être transportées à travers la membrane externe, de sorte que la composition en protéines diffère dans l'espace intermembranaire par rapport à celle du cytosol. Le cytochrome c est ainsi l'une des protéines situées dans l'espace intermembranaire mitochondrial[11].
Membrane interne
La membrane mitochondriale interne est sensiblement moins perméable que la membrane externe et ne contient pas de porines. Pratiquement toutes les molécules et les ions ont besoin d'un transporteur membranaire pour traverser la membrane interne. Les protéines sont transportées par les complexes translocase de la membrane interne (en) (TIM) ou par la protéine Oxa1[10]. Ainsi, le complexe TIM 23 permet l'entrée de protéines situées dans l'espace intermembranaire dans la membrane interne et dans la matrice mitochondriale. Le complexe TIM 22 permet l'insertion des protéines dans la membrane interne et notamment des protéines à plusieurs domaines transmembranaires. Les protéines Oxa permettent la sortie de la matrice pour certaines protéines d'origine mitochondriale.
La membrane interne contient plus de 151 polypeptides différents, elle héberge environ 1/8e de toutes les protéines d'une mitochondrie et possède un rapport pondéral protéines/phospholipides supérieur à 3:1, ce qui correspond à environ une protéine pour 15 molécules de phospholipides. La membrane interne possède notamment un phospholipide double, la cardiolipine[13], renfermant quatre acides gras au lieu de deux, ce qui pourrait contribuer à accroître l'imperméabilité de cette membrane ; ce phospholipide particulier a été découvert en 1942 dans des cœurs de bovins — d'où son nom — et est généralement caractéristique des membranes mitochondriales et des membranes plasmiques bactériennes. Les protéines de la membrane mitochondriale interne assurent essentiellement cinq types de fonctions physiologiques :
- réactions d'oxydoréduction de la phosphorylation oxydative ;
- production d'ATP à partir d'ADP par l'ATP synthase ;
- transporteurs membranaires assurant et régulant la circulation des métabolites de et vers la matrice mitochondriale ;
- système d'importation des protéines ;
- fusion (en) et fission des mitochondries (en).
Crêtes
La membrane interne forme des invaginations qui apparaissent au microscope électronique sous forme de replis appelés crêtes. Ces crêtes augmentent la surface de la membrane interne selon un facteur variable d'un type cellulaire à l'autre, environ d'un facteur 5 pour les mitochondries d'hépatocytes, voire davantage pour les mitochondries de myocytes. Elles ne sont pas distribuées au hasard, mais résultent au contraire d'invaginations susceptibles d'influencer les propriétés chimiosmotiques générales de l'organite[14]. C'est à leur niveau que ce concentrent de petits corps sphériques appelés particules F1 ou oxysomes. On y trouve en particulier les enzymes de la chaîne respiratoire, ainsi que l'ATP synthase sous la forme de complexes Fo-F1 visibles au microscope électronique comme des protubérances internes.
Matrice
La matrice mitochondriale est l'espace inclus dans la membrane mitochondriale interne. Elle renferme environ les deux tiers du total des protéines de la mitochondrie. Elle joue un rôle déterminant dans la production d'ATP avec l'aide de l'ATP synthase incluse dans la membrane interne. Elle contient un mélange très concentré de centaines d'enzymes différentes (principalement impliquées dans la dégradation des acides gras et du pyruvate), de ribosomes spécifiques aux mitochondries, d'ARN de transfert et plusieurs copies de l'ADN du génome mitochondrial.
Les mitochondries possèdent leur propre génome ainsi que l'équipement enzymatique nécessaire pour réaliser leur propre biosynthèse des protéines. La séquence du génome mitochondrial humain se compose de 16 569 paires de bases encodant 37 gènes[15] : 22 ARN de transfert, 2 ARN ribosomique et 13 polypeptides. Les 13 peptides mitochondriaux humains sont intégrées à la membrane mitochondriale interne avec des protéines encodées par des gènes situés dans le noyau de la cellule.
Origine

1. Nucléole ;
2. Noyau ;
3. Ribosomes ;
4. Vésicule ;
5. Réticulum endoplasmique rugueux (ou granuleux) (REG) ;
6. Appareil de Golgi ;
7. Cytosquelette ;
8. Réticulum endoplasmique lisse ;
9. Mitochondries ;
10. Vacuole ;
11. Cytosol ;
12. Lysosome ;
13. Centrosome (constitué de deux centrioles) ;
14. Membrane plasmique.
Une mitochondrie ne peut provenir que de la croissance et de la division d'une autre mitochondrie déjà existante. Normalement, avant la division cellulaire, la mitochondrie double sa masse puis se scinde en deux. Cette division débute par l'apparition d'un sillon de division sur la membrane interne. Elle a lieu pendant toute l'interphase et nécessite l'intervention de la protéine DRP1 (voisine de la dynamine). La réplication de l'ADN mitochondrial n'est pas limitée à la phase S du cycle cellulaire. Le nombre de mitochondries par cellule est régulé par l'activité cellulaire. Par exemple, une cellule musculaire au repos contient 5 à 10 fois moins de mitochondries qu'une cellule musculaire activée en permanence.
Le fait que la mitochondrie possède son ADN propre, comme les chloroplastes, indique une origine exogène : il est maintenant admis que les mitochondries proviennent de l'endosymbiose d'une α-protéobactérie il y a environ 2 milliards d'années. La théorie endosymbiotique de l'origine des mitochondries a été développée et argumentée par Lynn Margulis dès 1966, puis elle a été appuyée par la découverte de l'ADN spécifique des mitochondries en 1980. Il semble qu'au cours de l'évolution l'ADN originel de la bactérie ait subi diverses évolutions, perdu un grand nombre de gènes, parfois transférés dans l'ADN de la cellule hôte. Parallèlement à ce report de la synthèse de certaines protéines vers l'hôte, ce dernier a développé un arsenal de translocases, enzymes permettant le transfert de ces protéines vers la matrice mitochondriale.
Génome mitochondrial
Selon la théorie endosymbiotique, les mitochondries possèderaient une origine monophylétique unique. Une cellule de procaryote primitive aurait intégré un endosymbiote il y a environ 1,5 à 2 milliards d’années, lorsque l’atmosphère primitive s’est enrichie en oxygène[16],[17]. Les études phylogénétiques indiquent que cet endosymbiote est apparenté aux alphaprotéobactéries, le plus proche parent de la mitochondrie connu actuellement étant Rickettsia prowazekii, un parasite intracellulaire obligatoire[16] (c'est-à-dire une bactérie ne pouvant survivre, se développer et se reproduire qu'à l'intérieur des cellules de son hôte, en utilisant les ressources de ces dernières). Au cours de l’évolution, la majorité des gènes de l’endosymbiote originel auraient été perdus ou bien transférés vers le noyau de la cellule d'eucaryote hôte[17],[18]. En effet, les nombreux pseudogènes mitochondriaux présents dans le génome attestent d’un processus de transfert tout au long de l’évolution[19],[20].
Le matériel génétique (ADN mitochondrial) de la mitochondrie (qui est la seule partie des cellules animales à posséder son propre ADN, en plus du noyau) est souvent utilisé dans les recherches phylogénétiques. Le génome mitochondrial (ADNmt) humain est circulaire, ne possède pas d'introns, et est composé de 16 569 paires de bases, dont 13 cistrons codant des ARN messagers, 22 gènes codant des ARN de transfert, et 2 gènes codant des ARN ribosomiques.
Le génome mitochondrial peut être très différent d'une espèce à l'autre, il est extrêmement dynamique, et est souvent hétéroplasmique, c'est-à-dire que différentes formes coexistent au sein de la même cellule. Il peut être trouvé sous forme circulaire ou linéaire, double ou simple brin. Ces différentes formes sont, entre autres, les produits de la réplication du génome mitochondrial par un mécanisme de cercle roulant, mais aussi d'un mécanisme de réplication recombinaison-dépendant, similaire à la réplication du phage T4. Les génomes mitochondriaux sont habituellement représentés sous forme circulaire, le « cercle maître » qui correspond à la molécule décrivant le mieux le génome.
Les ribosomes mitochondriaux ou mitoribosomes sont différents des ribosomes de la cellule : ils sont plus petits (70S au lieu de 80S).
Le code génétique employé pour la synthèse des protéines peut être différent de celui utilisé dans les synthèses cytosoliques. Chez les vértébrés 4 codons sur 64 ont une signification différente, dont le codon UGA qui est transcrit dans le cytosol en codon stop mais dans la matrice UGA est transcrit en tryptophane (Trp/W), AGG et AGA codent un codon STOP au lieu d'une arginine (Arg/R) et AUA code la méthionine (Met/M) au lieu de l'isoleucine (Ile/I). L'ADN mitochondrial peut aussi se répliquer.
Chez les animaux, lors de la reproduction sexuée, les mitochondries du spermatozoïde pourraient passer dans l'ovocyte, mais le nombre de mitochondries ainsi transférées reste très faible en comparaison de celles déjà présentes dans l'ovocyte. Autrement dit, la quasi-totalité des mitochondries de la cellule-œuf provient du gamète femelle. L'étude de l'ADN mitochondrial humain permet donc de retracer les relations généalogiques entre les individus seulement selon la voie maternelle. Certaines études ont ainsi pu décrire un génome mitochondrial ancestral duquel descendraient tous les génomes mitochondriaux de l'humanité. L'individu femelle supposé qui portait ce génome a été dénommé Ève mitochondriale. Ce terme biblique reste toutefois trompeur, il est en effet très peu probable que l'humanité ait un unique ancêtre féminin et de récentes études, prouvant le transfert de mitochondries provenant des spermatozoïdes lors de la fécondation, remettent en cause cette théorie.
Protéome mitochondrial
Le protéome mitochondrial est l'ensemble des protéines présentes dans les mitochondries d'une cellule eucaryote à un moment donné. Le protéome est un ensemble dynamique défini dans le temps (moment considéré : stade de développement, matin ou soir) et dans l'espace (échantillon considéré : cellule, tissu, organisme). Pour décrire l'ensemble des protéines pouvant être présentes dans une mitochondrie à un moment quelconque de la vie de l'organisme, on utilisera le terme de protéome total.
Le protéome mitochondrial est composé de protéines produites dans les mitochondries et codées dans le génome mitochondrial, et de protéines produites dans le cytoplasme et codées dans le génome nucléaire. La plupart des complexes enzymatiques (exemple : ATP-synthase) sont formés par la juxtaposition de polypeptides synthétisés dans la mitochondrie et dans le cytosol (le fluide interne de la cellule).
Bien que le les mitochondries soient les descendantes de bactéries, les protéines de leur protéome ne sont pas toutes d'origine bactérienne, Ainsi chez la levure 50 à 60 % des protéines mitochondriales ont des homologues chez les procaryotes alors que 40 à 50 % n’en ont pas[17].
Il est intéressant de noter que c'est grâce à des associations des protéines de kinésine et de dynéine à des microtubules que la mitochondrie est capable de mouvement.
Protéines mitochondriales codées par le génome mitochondrial
Suivant les organismes 1 à 10 % des protéines mitochondriales sont directement synthétisées dans la matrice par les mitoribosomes, à partir de l'ADN mitochondrial.
Protéines mitochondriales codées par le génome nucléaire
Les protéines mitochondriales possédant un homologue procaryote résultent probablement du transfert des gènes de l’endosymbionte vers le nucléaire tandis que les protéines non homologues à des protéines procaryotes résultent d’un phénomène « d’enrichissement » du protéome mitochondrial par de nouvelles protéines et donc de nouvelles fonctions[16].
Les protéines mitochondriales codées par le génome nucléaire (ou protéines mitochondriales nucléaires) sont importées à l'intérieur de la matrice mitochondriale par différents mécanismes possibles :
- des complexes d'importation (3 sur la membrane interne, 2 sur la membrane externe);
- un peptide signal (environ 15 à 30 acides aminés) en position N-terminale de la protéine qui permet sa reconnaissance et son importation dans la mitochondrie[21],[22] ;
- grâce à un apport énergétique.
Chez l'Humain
La taille du protéome mitochondrial humain est estimée à plus d’un millier de protéines, dont environ 1 % codées par le génome mitochondrial (13 protéines) [23], dont actuellement la moitié est identifiée[24],[25]. Seules 13 protéines sont codées par l’ADN mitochondrial, vestige du génome de l’endosymbionte. Toutes les autres protéines sont codées par le génome nucléaire.
Fonctionnement
Elle est considérée comme la « centrale énergétique » de la cellule, car c'est là que se déroulent les dernières étapes du cycle respiratoire qui convertit l'énergie des molécules organiques issues de la digestion (glucose) en énergie directement utilisable par la cellule (l'ATP). En cas d'absence d'oxygène la cellule utilise la fermentation dans le cytoplasme pour produire l'énergie nécessaire à son fonctionnement, mais c'est un système bien moins efficace, qui dégrade le substrat de façon incomplète. L'augmentation de la concentration en ions H+ dans les cellules musculaires est une des raisons de la fatigue après une activité intense. En effet, ces ions H+ changent le pH intracellulaire et modifient de fait les conditions de fonctionnement enzymatiques de la cellule qui ne peut plus travailler correctement.
C'est dans la mitochondrie que se déroulent les deux dernières phases de la respiration cellulaire : le cycle de Krebs (dans la matrice) et la chaîne de transport d'électrons (au niveau de la membrane interne). En effet, la production d'ATP comporte 3 principales étapes :
- La glycolyse est la première étape. Elle se déroule dans le cytoplasme cellulaire.
- La deuxième étape est la production d'Acétyl-CoA dans la mitochondrie.
- La troisième et dernière étape est la phosphorylation oxydative.
Au cours de ces 3 étapes, via le cycle de Krebs (donc en condition d'aérobiose), la mitochondrie permet, à partir d'une molécule de glucose, la production théorique de 36 ou 38 molécules d'ATP(cela dépend de la navette utilisée pour transporter le NAD de la glycolyse) — en pratique, le rendement est un peu moins élevé, voisin d'une trentaine de molécules d'ATP par molécule de glucose oxydée, certaines études donnant la valeur de 29,85 ATP/glucose[26].
Les mitochondries participent à l'apoptose (mort cellulaire) avec le cytochrome C. De plus, elles ont aussi une fonction de concentration et de stockage des ions calcium, sodium et potassium où ils sont stockés sous forme de granules opaques. On trouve également de l'or, du fer et de l'osmium.
Poisons mitochondriaux
| Cibles des poisons | Poisons |
|---|---|
| complexe I | Roténone ; Barbituriques ; Dérivés mercuriels |
| complexe II | Malonate (acide malonique) |
| complexe III | Antimycine |
| complexe IV | Monoxyde d'azote ; Cyanure ; Monoxyde de carbone |
| complexe V (F0/F1ATPase) | Oligomycine ; Aurovertine |
| échangeur ATP/ADP | Atractyloside ; Acide bongkrékique |
| perméabilité de la membrane interne | Dinitrophénol ; Valinomycine |
Certains poisons ont pour rôle non pas d'empêcher les différents complexes de fonctionner, c'est-à-dire que les transferts d'électron de la chaîne respiratoire sont effectués mais ces protéines, les découplants ou UCP vont court-circuiter le complexe V (ATP synthase) en créant un canal à travers la membrane interne. Ce pore permet aux protons de passer de l'espace inter-membranaire vers la matrice dans le sens de leur gradient, ce qui se traduit par un dégagement de chaleur mais aucune production d'ATP. On peut citer ici l'exemple du dinitrophénol.
Maladies mitochondriales
- Myopathies
- Maladies neurodégénératives
- Ataxie de Friedreich : Maladie touchant la frataxine (Protéine mitochondriale impliquée dans l'imperméabilité des membranes au fer).
- Neuropathie optique de Leber
Historique
En 1857, Kölliker décrit les aspects de la mitochondrie dans le muscle. En 1890, Altmann décrit une technique de coloration des mitochondries qu'il appelle bioblastes et postule leur autonomie métabolique et génétique. En 1937, un scientifique allemand, Hans Adolf Krebs, élabore un modèle de voie métabolique connu sous le nom de cycle de Krebs qui se déroule, chez les eucaryotes, dans les mitochondries. En 1940-43, Claude isole les mitochondries dans des cellules du foie. En 1948-50, Kennedy et Lehninger montrent que le cycle de Krebs, la β-oxydation et la phosphorylation oxydative ont tous lieu dans les mitochondries. En 1978, Peter Mitchell obtient le prix Nobel pour sa théorie chimiosmotique. En 1981, Anderson et son équipe découvrent la structure génétique de l’ADN mitochondrial humain. Finalement, Boyer et Walker obtiennent également le prix Nobel pour leurs études sur la structure et le fonctionnement de l'ATP synthase.
Notes et références
- ↑ (en) Katrin Henze1 et William Martin, « Evolutionary biology: essence of mitochondria », Nature, vol. 426, no 6963, , p. 127-128 (PMID 14614484, DOI 10.1038/426127a, lire en ligne)
- ↑ (en) Heidi M. McBride, Margaret Neuspiel et Sylwia Wasiak, « Mitochondria: More Than Just a Powerhouse », Current Biology, vol. 16, no 14, , R551-R560 (PMID 16860735, DOI 10.1016/j.cub.2006.06.054, lire en ligne)
- ↑ (en) Ann Gardner et Richard G. Boles, « Is a "Mitochondrial Psychiatry" in the Future? A Review », Current Psychiatry Reviews, vol. 1, no 3, , p. 255-271 (DOI 10.2174/157340005774575064, lire en ligne)
- ↑ (en) Edward J. Lesnefsky, Shadi Moghaddas, Bernard Tandler, Janos Kerner et Charles L. Hoppel, « Mitochondrial Dysfunction in Cardiac Disease: Ischemia–Reperfusion, Aging, and Heart Failure », Journal of Molecular and Cellular Cardiology, vol. 33, no 6, , p. 1065-1089 (PMID 11444914, DOI 10.1006/jmcc.2001.1378, lire en ligne)
- ↑ (en) Steven W. Taylor, Eoin Fahy, Bing Zhang, Gary M. Glenn, Dale E. Warnock, Sandra Wiley, Anne N. Murphy, Sara P. Gaucher, Roderick A. Capaldi, Bradford W. Gibson et Soumitra S. Ghosh, « Characterization of the human heart mitochondrial proteome », Nature Biotechnology, vol. 21, no 3, , p. 281-286 (PMID 12592411, DOI 10.1038/nbt793, lire en ligne)
- ↑ (en) Jun Zhang, Xiaohai Li, Michael Mueller, Yueju Wang, Chenggong Zong, Ning Deng, Thomas M. Vondriska, David A. Liem, Jeong-In Yang, Paavo Korge, Henry Honda, James N. Weiss, Rolf Apweiler and Peipei Ping, « Systematic characterization of the murine mitochondrial proteome using functionally validated cardiac mitochondria », Proteomics, vol. 8, no 8, , p. 1564-1575 (PMID 18348319, PMCID 2799225, DOI 10.1002/pmic.200700851, lire en ligne)
- ↑ (en) Jun Zhang, David A. Liem, Michael Mueller, Yueju Wang, Chenggong Zong, Ning Deng, Thomas M. Vondriska, Paavo Korge, Oliver Drews, W. Robb MacLellan, Henry Honda, James N. Weiss, Rolf Apweiler et Peipei Ping, « Altered Proteome Biology of Cardiac Mitochondria Under Stress Conditions », Journal of Proteome, vol. 7, no 6, , p. 2204-2214 (PMID 18484766, PMCID 3805274, DOI 10.1021/pr070371f, lire en ligne)
- ↑ (en) G. E. Andersson, Olof Karlberg, Björn Canbäck et Charles G. Kurland, « On the origin of mitochondria: a genomics perspective », Philosophical Transactions B, vol. 358, no 1429, , p. 165-177 (PMID 12594925, PMCID 1693097, DOI 10.1098/rstb.2002.1193, lire en ligne)
- ↑ Pierre Cau, Raymond Seïte et Andrée Robaglia-Schlupp, Cours de Biologie cellulaire, Ellipses,
- 1 2 (en) Johannes M. Herrmann et Walter Neupert, « Protein transport into mitochondria », Current Opinion in Microbiology, vol. 3, no 2, , p. 210-214 (PMID 10744987, DOI 10.1016/S1369-5274(00)00077-1, lire en ligne)
- 1 2 (en) J. E. Chipuk, L. Bouchier-Hayes et D. R. Green, « Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario », Cell Death and Differentiation, vol. 13, no 8, , p. 1396-1402 (PMID 16710362, DOI 10.1038/sj.cdd.4401963, lire en ligne)
- ↑ (en) Teruo Hayashi, Rosario Rizzuto, Gyorgy Hajnoczky et Tsung-Ping Su, « MAM: more than just a housekeeper », Trends in Cell Biology, vol. 19, no 2, , p. 81-88 (PMID 19144519, PMCID 2750097, DOI 10.1016/j.tcb.2008.12.002, lire en ligne)
- ↑ (en) Jeanie B McMillin et William Dowhan, « Cardiolipin and apoptosis », Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, vol. 1585, no 2-3, , p. 97-107 (PMID 12531542, DOI 10.1016/S1388-1981(02)00329-3, lire en ligne)
- ↑ (en) Carmen A. Mannella, « Structure and dynamics of the mitochondrial inner membrane cristae », Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1763, no 5-6, , p. 542-548 (PMID 16730811, DOI 10.1016/j.bbamcr.2006.04.006, lire en ligne)
- ↑ (en) S. Anderson, A. T. Bankier, B. G. Barrell, M. H. L. de Bruijn, A. R. Coulson, J. Drouin, I. C. Eperon, D. P. Nierlich, B. A. Roe, F. Sanger, P. H. Schreier, A. J. H. Smith, R. Staden et I. G. Young, « Sequence and organization of the human mitochondrial genome », Nature, vol. 290, no 5806, , p. 457-465 (PMID 7219534, DOI 10.1038/290457a0, Bibcode 1981Natur.290..457A, lire en ligne)
- 1 2 3 (en) S.G. Andersson, A. Zomorodipour, J.O. Andersson, T. Sicheritz-Ponten, U.C. Alsmark, R.M. Podowski, A.K. Naslund, A.S. Eriksson, H.H. Winkler & C.G. Kurland, « The genome sequence of Rickettsia prowazekii and the origin of mitochondria », Nature, vol. 396, no 6707, , p. 133–140 (PMID 9823893, DOI 10.1038/24094)
- 1 2 3 (en) M.W. Gray, G. Burger & B.F. Lang, « The origin and early evolution of mitochondria », Genome Biology, vol. 2, no 6, , reviews1018.1–1018.5 (PMID 11423013, DOI 10.1186/gb-2001-2-6-reviews1018)
- ↑ (en) C.G. Kurland & S.G. Andersson, « Origin and evolution of the mitochondrial proteome », Microbiology and Molecular Biology Reviews, vol. 64, no 4, , p. 786–820 (PMID 11104819)
- ↑ (en) Y. Tourmen, O. Baris, P. Dessen, C. Jacques, Y. Malthiery, & P. Reynier, « Structure and chromosomal distribution of human mitochondrial pseudogenes », Genomics, vol. 80, no 1, , p. 71–77 (PMID 12079285, DOI 10.1006/geno.2002.6798)
- ↑ (en) Woischnik, M. and C.T. Moraes, « Pattern of organization of human mitochondrial pseudogenes in the nuclear genome », Genome Research, vol. 12, no 6, , p. 885–893 (PMID 12045142, DOI 10.1101/gr.227202)
- ↑ (en) Rusch, S.L. & D.A. Kendall, « Protein transport via amino-terminal targeting sequences: common themes in diverse systems », Molecular Membrane Biology, vol. 12, no 4, , p. 295–307 (PMID 8747274, DOI 10.3109/09687689509072431)
- ↑ (en) G. Schatz & B. Dobberstein, « Common principles of protein translocation across membranes », Science, vol. 271, no 5255, , p. 1519–1526 (PMID 8599107, DOI 10.1126/science.271.5255.1519)
- ↑ (en) M.F. Lopez, B.S. Kristal, E. Chernokalskaya, A. Lazarev, A.I. Shestopalov, A. Bogdanova, & M. Robinson, « High-throughput profiling of the mitochondrial proteome using affinity fractionation and automation », Electrophoresis, vol. 21, no 16, , p. 3427–3440 (PMID 11079563)
- ↑ (en) C. Andreoli, H. Prokisch, K. Hortnagel, J.C. Mueller, M. Munsterkotter, C. Scharfe, & T. Meitinger, « MitoP2, an integrated database on mitochondrial proteins in yeast and man », Nucleic Acids Research, vol. 32, no Database issue, , D459–D462 (PMID 14681457, DOI 10.1093/nar/gkh137)
- ↑ (en) D. Cotter, P. Guda, E. Fahy, & S. Subramaniam, « MitoProteome: mitochondrial protein sequence database and annotation system », Nucleic Acids Research, vol. 32, no Database issue, , D463–D467 (PMID 14681458, DOI 10.1093/nar/gkh048)
- ↑ (en) P. R. Rich, « The molecular machinery of Keilin's respiratory chain », Biochemical Society Transactions, vol. 31, no Pt 6, , p. 1095-1105 (lire en ligne) DOI:10.1042/BST0311095
Articles connexes
- ATP synthase
- Découplage mitochondrial
- Respiration cellulaire
- Chaîne respiratoire
- Section Production d'énergie dans électrophysiologie
- Génome mitochondrial
- Génome mitochondrial humain
Liens externes
- [vidéo] (27 minutes) : La génétique de la mitochondrie chez les paramécies
- [vidéo] (4 minutes) : Cours : La mitochondrie
- Les mitochondries - eBiologie.fr
 Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire