
{kind=link}

Prion
Contexte des ??coles Wikip??dia
Ce contenu de Wikipedia a ??t?? s??lectionn?? par SOS Enfants d'aptitude dans les ??coles ?? travers le monde. M??res SOS chaque regard apr??s une une famille d'enfants parrain??s .
| Maladies ?? prions (EST) | |
|---|---|
| les ressources de classification et externes | |
 "Trous" sont caract??ristiques microscopiques dans des coupes de tissus prions affect??, ce qui provoque le tissu de d??velopper une architecture "spongieux". | |
| CIM 10 | Un 81 |
| CIM 9 | 046 |
Un prion ( / p r Je ɒ n /) Sous la forme de la tremblante (PrP Sc) est un agent infectieux compos?? de prot??ine dans un forme mal repli??e. Ce est l'id??e centrale de la Prion hypoth??se, qui reste d??battue. Ce serait, contrairement ?? tous les autres agents infectieux connus ( virus / bact??ries / champignons / parasite) qui doit contenir des acides nucl??iques (soit ADN , ARN, ou les deux). Le mot prion, invent?? en 1982 par Stanley Prusiner, est d??riv?? de la prot??ine de mots et l'infection. Les prions sont responsables de la les enc??phalopathies spongiformes transmissibles dans une vari??t?? de mammif??res , y compris enc??phalopathie spongiforme bovine (l'ESB, ??galement connu sous le nom de ??maladie de la vache folle") dans les bovins et La maladie de Creutzfeldt-Jakob (MCJ) chez l'homme. Toutes les maladies ?? prions connus affectent la structure du cerveau ou autre tissu neural et tous sont actuellement incurable et universellement fatale.
Prions propagent en transmettant un Etat des prot??ines mal repli??es. Quand un prion entre un organisme sain, il induit prot??ines existantes, correctement repli??es pour convertir en la forme de prion associ??e ?? la maladie; les actes ?? prions comme un mod??le pour guider le mauvais repliement des prot??ines en plus sous forme de prion. Ces prions nouvellement form??es peuvent ensuite passer ?? convertir plus de prot??ines elles-m??mes; ce qui d??clenche une r??action en cha??ne qui produit de grandes quantit??s de la forme prion. Tous les prions connus induisent la formation d'une amylo??de fois, dans lequel les polym??rise de prot??ines dans un agr??gat compos?? de serr??s feuillets b??ta. agr??gats amylo??des fibrilles sont, de plus en plus ?? leurs extr??mit??s, et de se r??pliquer lorsque la rupture provoque deux extr??mit??s de croissance pour devenir quatre extr??mit??s en croissance. Le p??riode d'incubation des maladies ?? prion est d??termin?? par la croissance exponentielle de d??bit associ?? ?? la r??plication des prions, qui est un ??quilibre entre la croissance lin??aire et la rupture des agr??gats. (Notez que la propagation du prion d??pend de la pr??sence de la prot??ine normalement repli??e dans laquelle le prion peut induire mauvais repliement;. Les animaux qui ne expriment pas la forme normale de la prot??ine prion ne peut se d??velopper ni transmettre la maladie)
Cette structure modifi??e est extr??mement stable et se accumule dans les tissus infect??s, entra??nant des l??sions tissulaires et la mort cellulaire. Cela signifie que la stabilit?? structurelle des prions sont r??sistants ?? d??naturation par des agents chimiques et physiques, faisant ??vacuation et le confinement de ces particules difficiles. Les prions sont de diff??rentes souches, chacune avec une structure l??g??rement diff??rente, et la plupart du temps, les souches se reproduisent vrai. la r??plication des prions est n??anmoins soumis ?? occasionnelle ??pimutation puis la s??lection naturelle comme les autres formes de r??plication. Cependant, le nombre de possibles souches de prion distinctes est probablement beaucoup plus petit que le nombre de s??quences d'ADN possibles, donc l'??volution a lieu dans un espace limit??.
Toutes les maladies ?? prions mammif??res connues sont caus??es par la dite prot??ine prion, PrP. Le endog??ne, bien pli??e, forme est not??e PrP C (C ou C ommune ellular) tandis que la forme mal repli??e de la maladie li??e est not??e PrP Sc (pour Sc rapie, apr??s une premi??re des maladies li??es aux prions et la neurod??g??n??rescence.) La structure pr??cise du prion est pas connue, mais ils peuvent ??tre form??s en combinant PrP C, l'acide polyad??nylique, et les lipides dans un Mauvais repliement des prot??ines cyclique Amplification (PMCA) r??action.
Prot??ines montrant de type prion comportement sont ??galement pr??sents dans certains champignons , qui a ??t?? utile pour aider ?? comprendre les prions de mammif??res. Prions fongiques ne semblent pas provoquer la maladie chez leurs h??tes.
D??couverte
Pendant les ann??es 1960, deux chercheurs bas??s ?? Londres, biologiste de rayonnement Tikvah Alper et math??maticien John Stanley Griffith d??velopp?? l'hypoth??se que certains les enc??phalopathies spongiformes transmissibles sont provoqu??es par un agent infectieux constitu??s exclusivement de prot??ines. Alper et Griffith voulu pour tenir compte de la d??couverte selon laquelle l'agent infectieux myst??rieuse provoquant les maladies tremblante et La maladie de Creutzfeldt-Jakob r??sist?? rayonnement ionisant. (A ionisants single "frapp??" d??truit normalement toute une particule infectieuse, et la dose n??cessaire de frapper la moiti?? des particules d??pend de la taille des particules. Les donn??es sugg??rent que l'agent infectieux ??tait trop petit pour ??tre un virus.)
Francis Crick a reconnu l'importance potentielle de la prot??ine seule hypoth??se Griffith pour la propagation de la tremblante dans la deuxi??me ??dition de son " Dogme central de la biologie mol??culaire "(1970): tout en affirmant que la circulation de l'information s??quence d'une prot??ine ??, ou ?? partir de prot??ines ?? l'ARN et l'ADN ??tait" emp??ch?? ", il a not?? que l'hypoth??se de Griffith ??tait une contradiction potentielle (bien que ce ne ??tait pas promu par Griffith). L'hypoth??se r??vis??e a ??t?? formul??e plus tard, en partie, ?? accueillir transcription inverse (qui ?? la fois Howard Temin et David Baltimore d??couvert en 1970).
En 1982, Stanley Prusiner du Universit?? de Californie, San Francisco a annonc?? que son ??quipe avait purifi?? le prion infectieuse hypoth??tique, et que l'agent infectieux consist?? principalement d'une prot??ine sp??cifique - se ils ne ont pas r??ussi ?? isoler la prot??ine que deux ans apr??s l'annonce de Prusiner. Bien que l'agent infectieux a ??t?? nomm?? prion, la prot??ine sp??cifique du prion qui ??tait compos?? de est ??galement connu sous le nom ion Pr P rotein (PrP), bien que cette prot??ine peut se produire ?? la fois sous des formes infectieuses et non infectieuses. Prusiner a remport?? le Prix Nobel de physiologie ou m??decine en 1997 pour ses recherches sur les prions.
Structure
Isoformes
La prot??ine prion qui sont faites de (PrP) se trouve dans tout le corps, m??me chez les personnes et les animaux en bonne sant??. Cependant, la PrP trouv??e dans le mat??riel infectieux a une structure diff??rente et est r??sistant ?? prot??ases, les enzymes dans le corps qui peut normalement d??composer les prot??ines. La forme normale de la prot??ine est appel?? PrP C, tandis que la forme infectieuse est appel?? PrP Sc - C se r??f??re ?? ??cellulaire?? ou ??commun?? PrP, tandis que le Sc se r??f??re ?? ' la tremblante du mouton ??, une maladie ?? prion se produisant chez les ovins. Alors que la PrP C est structurellement bien d??finie, PrP Sc est certainement polydisperse et d??finie ?? un niveau relativement faible. PrP peut ??tre amen?? ?? se replier dans d'autres isoformes plus ou moins bien d??finis in vitro, et leur relation ?? la forme (s) qui sont pathog??nes in vivo ne est pas encore clair.
PrP C
PrP C est une prot??ine normale trouv?? sur le des membranes de cellules . Il poss??de 209 acides amin??s (chez l'homme), une Pont disulfure, une masse mol??culaire de 35-36 et un kDa principalement la structure en h??lice alpha. Plusieurs formes topologiques existent; une forme de la surface de la cellule par l'interm??diaire ancr??e glycolipide et deux formes transmembranaires. La prot??ine ne est pas normale sedimentable; ce qui signifie qu'il ne peut pas ??tre s??par?? par centrifugation techniques. Sa fonction est une question complexe qui continue d'??tre ??tudi??. PrP C lie le cuivre (II) des ions avec une haute affinit??. L'importance de cette d??couverte ne est pas clair, mais il concerne vraisemblablement ?? la structure ou la fonction PrP. PrP C est facilement dig??r?? par prot??inase K et peut ??tre lib??r?? de la surface cellulaire in vitro par l'enzyme phosphoinositide phospholipase C (PI-PLC), qui clive les glycophosphatidylinositol (GPI) de l'ancre glycolipidique. PrP a ??t?? rapport?? ?? jouer un r??le important dans l'adh??sion cellule-cellule et la signalisation intracellulaire in vivo, et peut donc ??tre impliqu?? dans la communication cellule-cellule dans le cerveau.
PrP Sc
Le infectieuse isoforme de PrP, appel??e PrP Sc, est capable de convertir les prot??ines normales PrPC dans l'isoforme infectieuse en changeant leur conformation, ou la forme; Ceci, ?? son tour, modifie la mani??re dont le interconnexion prot??ines. Bien que la structure 3D exacte de la PrPSc ne est pas connue, il a une plus grande proportion de β-feuille de la structure en place la normale α-h??lice structure. Agr??gations de ces isoformes anormales forme hautement structur??es fibres amylo??des, qui se accumulent pour former des plaques. On ne sait pas si ces agr??gats sont ?? l'origine de l??sions cellulaires ou sont tout simplement un effet secondaire du processus de la maladie sous-jacente. L'extr??mit?? de chaque fibre agit comme une matrice sur laquelle les mol??cules de prot??ines libres peuvent fixer, ce qui permet ?? la fibre de se d??velopper. Dans la plupart des cas, seuls les mol??cules de PrP avec une s??quence d'acides amin??s identique ?? la PrP Sc infectieux sont incorpor??s dans la fibre de croissance. Cependant, la transmission inter-esp??ces rares est ??galement possible. Dans un autre prion, Sup35p a ??t?? montr?? pour ??tre en mesure d'??tre incorpor?? dans les agr??gations existantes m??me si trois des cinq r??p??titions d'oligopeptides normalement pr??sents ont ??t?? supprim??s.
m??canisme de replication Prion

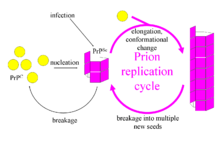
La premi??re hypoth??se qui a tent?? d'expliquer comment les prions r??pliquent de fa??on que la prot??ine est le mod??le d'h??t??rodim??res. Ce mod??le suppose que d'une seule mol??cule PrP Sc se lie ?? une seule mol??cule de PrP C et catalyse sa conversion en PrP Sc. Les deux mol??cules de PrP Sc viennent alors dehors et peuvent passer ?? convertir plus PrP C. Cependant, un mod??le de r??plication du prion doit expliquer ?? la fois la fa??on dont les prions se propagent, et pourquoi leur apparition spontan??e est si rare. Manfred Eigen a montr?? que le mod??le de h??t??rodim??re n??cessite PrP Sc ??tre un efficace extr??mement catalyseur, ce qui augmente la vitesse de la r??action de conversion d'un facteur de l'ordre de 10 15. Ce probl??me ne se pose pas si la PrPSc ne existe que dans des formes agr??g??es telles que amylo??de, o?? coop??rativit?? peut agir comme une barri??re ?? la conversion spontan??e. Qui plus est, malgr?? des efforts consid??rables, infectieuse monom??re PrP Sc n'a jamais ??t?? isol??.
Un autre mod??le suppose que PrP Sc ne existe que comme fibrilles, fibrilles et que se termine lie PrP C et le convertir en PrP Sc. Se il en ??tait tout, alors la quantit?? de prions augmenterait lin??aire, formant des fibrilles plus longues jamais. Mais la croissance exponentielle des deux PrP Sc et du la quantit?? de particules infectieuses est observ??e au cours de la maladie ?? prions. Ceci peut se expliquer en tenant compte de la rupture de fibrilles. Une solution math??matique du taux de croissance exponentielle r??sultant de la combinaison de la croissance des fibrilles et la rupture des fibrilles a ??t?? trouv??. Le taux de croissance exponentielle d??pend largement de la racine carr??e de la concentration PrP C. Le p??riode d'incubation est d??termin??e par le taux de croissance exponentielle, et des donn??es in vivo sur les maladies ?? prions dans souris transg??niques correspondent cette pr??diction. La m??me d??pendance de la racine carr??e est ??galement vu dans des exp??riences in vitro avec une grande vari??t?? de prot??ines amylo??des.
Le m??canisme de la r??plication du prion a des implications pour la conception de m??dicaments. Depuis la p??riode d'incubation des maladies ?? prions est si longue, un m??dicament efficace ne est pas n??cessaire d'??liminer toutes les prions, mais a simplement besoin de ralentir le taux de croissance exponentielle. Les mod??les pr??disent que le moyen le plus efficace pour atteindre cet objectif, utilisant un m??dicament ?? la dose la plus faible possible, est de trouver un m??dicament qui se lie ?? fibrilles extr??mit??s et les bloque de plus en plus loin.
Fonction PrP
Il a ??t?? propos?? que la neurod??g??n??rescence caus??e par des prions peut ??tre li??e ?? une fonction anormale de la PrP. Toutefois, la fonction physiologique de la prot??ine prion reste un sujet de controverse. Bien que les donn??es provenant d'exp??riences in vitro sugg??rent de nombreux r??les diff??rents, des ??tudes sur des souris knock-out PrP ont fourni des informations limit??es parce que ces animaux pr??sentent des anomalies mineures seulement. Dans une recherche r??cente effectu??e chez la souris, on a trouv?? que le clivage des prot??ines PrP dans les nerfs p??riph??riques provoque l'activation de r??paration de la my??line dans Les cellules de Schwann et que le manque de prot??ines PrP caus??s d??my??linisation dans ces cellules.
PrP m??moire et ?? long terme
Un examen de la preuve en 2005 sugg??r?? que PrP peut avoir une fonction normale dans l'entretien des la m??moire ?? long terme. En outre, une ??tude de 2004 a r??v??l?? que les souris d??pourvues de g??nes pour le spectacle de la prot??ine PrP cellulaire normale modifi?? hippocampique potentialisation ?? long terme.
PrP et les cellules souches renouvellement
Un article de l'Institut Whitehead pour la recherche biom??dicale 2006 indique que l'expression de la PrP sur les cellules souches est n??cessaire pour l'auto-renouvellement d'un organisme de la moelle osseuse. L'??tude a montr?? que tous ?? long terme les cellules souches h??matopo????tiques expriment PrP sur leur membrane cellulaire et que les tissus h??matopo????tiques avec des cellules souches PrP nulles expos??es sensibilit?? accrue ?? la d??pl??tion des cellules.
Maladie ?? prions
| Animal atteint (s) | Maladie |
|---|---|
| moutons , ch??vres | Tremblante |
| b??tail | Enc??phalopathie spongiforme bovine (ESB de), la maladie de la vache folle |
| vison | Enc??phalopathie transmissible du vison (TME) |
| le cerf de Virginie, le wapiti, le cerf mulet, l'orignal | La maladie du d??p??rissement chronique (MDC) |
| chat | Enc??phalopathie spongiforme f??line (ESF) |
| Nyala, oryx, grand koudou | Exotique ongul??s bovine (EUE) |
| autruche | Enc??phalopathie spongiforme (N'a pas ??t?? d??montr?? qu'il est transmissible.) |
| humain | La maladie de Creutzfeldt-Jakob (MCJ) |
| La maladie de Creutzfeldt-Jakob iatrog??ne (MCJi) | |
| Variante de la maladie de Creutzfeldt-Jakob (vMCJ) | |
| Familial maladie de Creutzfeldt-Jakob (fMCJ) | |
| Sporadique maladie de Creutzfeldt-Jakob (MCJ) | |
| Syndrome de Gerstmann-Str??ussler-Scheinker (GSS) | |
| L'insomnie fatale familiale (IFF) | |
| Kuru |
Les prions causent la maladie neurod??g??n??rative par agr??gation au niveau extracellulaire dans le syst??me nerveux central pour former des plaques appel??es amylo??de, ce qui perturbe la normale la structure des tissus. Cette perturbation est caract??ris?? par des ??trous?? dans le tissu avec l'architecture spongieuse r??sultante en raison de la la formation de vacuoles dans les neurones. Autres modifications histologiques comprennent astrogliose et l'absence d'un r??action inflammatoire. Tandis que le p??riode d'incubation de maladies ?? prions est g??n??ralement assez long, une fois les sympt??mes apparaissent la maladie progresse rapidement, menant ?? des l??sions c??r??brales et la mort. Neurod??g??n??ratives sympt??mes peuvent inclure convulsions, la d??mence, ataxie (??quilibre et la dysfonction de coordination), et les changements de comportement ou de la personnalit??.
Toutes les maladies ?? prions, connus collectivement appel??es enc??phalopathies spongiformes transmissibles (EST), sont incurable et fatale. Un vaccin a ??t?? d??velopp?? chez la souris, cependant, qui peuvent donner un aper??u de fournir un vaccin chez l'humain ?? r??sister aux infections ?? prions. En outre, en 2006 les scientifiques ont annonc?? qu'ils avaient g??n??tiquement le b??tail manque un g??ne n??cessaire ?? la production de prion - donc th??oriquement faire ?? l'abri de l'ESB, en se appuyant sur la recherche indiquant que les souris d??pourvues survenant normalement prot??ine prion sont r??sistantes ?? l'infection par la prot??ine tremblante du prion.
De nombreuses esp??ces de mammif??res diff??rentes peuvent ??tre affect??es par les maladies ?? prions, comme la prot??ine prion (PrP) est tr??s similaire dans tous les mammif??res. En raison de petites diff??rences dans la PrP entre diff??rentes esp??ces, il est inhabituel pour une maladie ?? prion ?? transmettre d'une esp??ce ?? une autre. La variante de la maladie ?? prions humaine maladie de Creutzfeldt-Jakob, cependant, est cens?? ??tre caus??e par un prion qui infecte typiquement bovins, causant Enc??phalopathie spongiforme bovine et est transmise par la viande infect??e.
Transmission
Il a ??t?? reconnu que les maladies ?? prions peuvent se produire de trois fa??ons diff??rentes: acquis, familial ou sporadique. On suppose souvent que la forme malade interagit directement avec la forme normale pour le faire modifier sa structure. Une id??e, l'hypoth??se "prot??ine X", qui est une prot??ine cellulaire qui n'a pas encore non identifi?? (prot??ine X) permet la conversion de la PrP C et PrP Sc en apportant une mol??cule de chacun des deux ensemble dans un complexe.
La recherche actuelle indique que la m??thode primaire de l'infection chez les animaux est l'ingestion. On pense que les prions peuvent ??tre d??pos??s dans l'environnement ?? travers les restes d'animaux morts et via l'urine, la salive et d'autres fluides corporels. Elles peuvent alors se attarder dans le sol par liaison ?? de l'argile et d'autres min??raux.
Une ??quipe de l'Universit?? de Californie de recherche, dirig??e par le prix Nobel Stanley Prusiner, a fourni des preuves de la th??orie que l'infection peut se produire ?? partir de prions dans le fumier. Et puisque le fumier est pr??sent dans de nombreux domaines entourant les r??servoirs d'eau, ainsi que de nombreux utilis??s sur les champs de culture, il soul??ve la possibilit?? d'une transmission ??tendue. Il a ??t?? rapport?? en Janvier 2011 que les chercheurs avaient d??couvert prions d'??pandage par voie a??rienne sur Les particules d'a??rosol, dans une exp??rience de l'exp??rimentation animale se concentrant sur l'infection de la tremblante chez les souris de laboratoire. Les donn??es pr??liminaires appuyant l'id??e que les prions peuvent ??tre transmises par l'utilisation de l'urine d??riv??s la gonadotrophine m??nopausique humaine, administr??e pour le traitement de infertilit??, a ??t?? publi?? en 2011.
St??rilisation
Particules infectieuses poss??dant acide nucl??ique sont tributaires ?? diriger leur r??plication continue. Les prions, cependant, sont infectieux par leur effet sur les versions normales de la prot??ine. St??rilisation prions requiert donc la d??naturation de la prot??ine ?? un ??tat o?? la mol??cule ne est plus capable d'induire le pliage anormal des prot??ines normales. Les prions sont g??n??ralement assez r??sistant ?? la prot??ases, la chaleur , rayonnement, et formol traitements, bien que leur infectivit?? peut ??tre r??duit par de tels traitements. La d??contamination de prion efficace repose sur l'hydrolyse ou la r??duction ou la destruction de la prot??ine structure tertiaire des prot??ines. Des exemples comprennent l'eau de Javel, soude caustique , et les d??tergents fortement acides tels que LpH. 134 ?? C (274 ?? F) pendant 18 minutes dans une vapeur sous pression autoclave peut ne pas ??tre suffisant pour d??sactiver l'agent de la maladie. st??rilisation ?? l'ozone est actuellement ?? l'??tude comme une m??thode potentiel de prion d??naturer et la d??sactivation. La renaturation d'un prion compl??tement d??natur?? ?? l'??tat infectieux n'a pas encore ??t?? atteint; Toutefois, les prions peuvent ??tre partiellement d??natur??es renatur??es ?? un ??tat infectieux sous certaines conditions artificielles.
L' Organisation mondiale de la sant?? recommande une des trois proc??dures suivantes pour la st??rilisation de tous les instruments chirurgicaux r??sistant ?? la chaleur pour se assurer qu'ils ne sont pas contamin??s par des prions:
- Plongez dans une casserole contenant 1N NaOH et la chaleur dans un autoclave ?? d??placement par gravit?? ?? 121 ?? C pendant 30 minutes; nettoyer; rincer ?? l'eau; puis ex??cuter des processus de st??rilisation de routine.
- Plongez dans l'univers 1N ou NaClO l'hypochlorite de sodium (20 000 parties par million de chlore actif) pendant 1 heure; transf??rer instruments ?? l'eau; la chaleur dans un autoclave ?? d??placement par gravit?? ?? 121 ?? C pendant 1 heure; nettoyer; puis ex??cuter des processus de st??rilisation de routine.
- Immerger dans du NaOH 1 N ou de l'hypochlorite de sodium (20 000 parties par million de chlore actif) pendant 1 heure; enlever et rincer ?? l'eau, puis les transf??rer dans un bac ouvert et de la chaleur dans une gravit??-d??placement (121 ?? C) ou dans un poreuse-charge (134 ?? C) autoclave pendant 1 heure; nettoyer; puis ex??cuter des processus de st??rilisation de routine.
D??bat
Que les prions sont l'agent qui provoque la maladie ou simplement un sympt??me caus?? par un autre agent est encore d??battue par une minorit?? de chercheurs. Les sections suivantes d??crivent plusieurs hypoth??ses: certaines se rapportent ?? la composition de l'agent infectieux (prot??ine seule, prot??ine avec d'autres composants, un virus, ou autre), tandis que d'autres se rapportent ?? son m??canisme de reproduction.
Protein-seule hypoth??se
Avant la d??couverte des prions, on a pens?? que tout pathog??nes utilis??s acides nucl??iques pour diriger leur r??plication. La "prot??ine seule hypoth??se" indique que la structure de la prot??ine peut se r??pliquer sans l'utilisation d'acide nucl??ique. Ce ??tait d'abord controvers??e car elle contredit la dogme central de la biologie mol??culaire, qui d??crit un acide nucl??ique comme la forme r??plicative centrale d'informations.
Preuve en faveur d'une hypoth??se de prot??ines ne inclut:
- Aucune particule de virus, des bact??ries ou des champignons ont ??t?? concluante associ??e aux maladies ?? prion, bien Saccharomyces cerevisiae est connu pour ??tre associ?? ?? des prions infectieux, mais non l??tales, telles que Sup35p.
- Aucun acide nucl??ique a ??t?? concluante associ??e ?? l'infectiosit??; l'agent est r??sistant aux rayonnements ultraviolets et nucl??ases.
- Pas de r??ponse immunitaire ?? l'infection.
- PrP Sc exp??rimentalement transmis entre une esp??ce ?? l'autre se traduit par PrP Sc avec la s??quence d'amino-acide de l'esp??ce r??ceptrice, ce qui sugg??re que la r??plication de l'agent donneur ne se produit pas.
- Maladie ?? prion familiale survient dans les familles avec une mutation dans le g??ne PrP, et les souris avec des mutations PrP d??velopper la maladie ?? prion, malgr?? des conditions contr??l??es o?? la transmission est emp??ch??e.
- Animaux d??pourvus de PrP C ne contractent pas la maladie ?? prion.
Les facteurs g??n??tiques
Un g??ne de la prot??ine normale a ??t?? identifi??: le G??ne PRNP. Dans tous les cas h??r??ditaires de la maladie ?? prion, une mutation dans le g??ne PRNP. Beaucoup de diff??rentes mutations du g??ne PRNP ont ??t?? identifi??s et ces prot??ines sont plus susceptibles de se replier en prion anormale. Bien que cette d??couverte met un trou dans l'hypoth??se g??n??rale de prion, que les prions peuvent seulement des prot??ines agr??g??es des acides amin??s identique maquillage. Ces mutations peuvent se produire tout au long du g??ne. Certaines mutations impliquent l'expansion de la r??gion de r??p??tition d'octapeptide ?? l'extr??mit?? N-terminale de la PrP. D'autres mutations qui ont ??t?? identifi??s comme une cause de la maladie du prion h??rit?? se produisent ?? des positions 102, 117 et 198 (ESG), 178, 200, 210 et 232 (MCJ) et 178 ( Insomnie fatale familiale, FFI). La cause de la maladie ?? prions peuvent ??tre sporadiques, g??n??tique et infectieuse , ou une combinaison de ces facteurs. Par exemple, afin d'avoir la tremblante du mouton, ?? la fois un agent infectieux et un g??notype sensible doivent ??tre pr??sents.
Hypoth??se multi-composants
Malgr?? beaucoup d'efforts, des titres significatifs de l'infectiosit?? des prions ne ont jamais ??t?? produites par le repliement des mol??cules de PrP pures, soulevant le doute sur la validit?? de l'hypoth??se ??prot??ine seulement??. En outre, l'hypoth??se de la ??prot??ine seulement?? ne parvient pas ?? fournir une explication mol??culaire de la capacit?? des souches de prions pour cibler des zones sp??cifiques du cerveau dans des mod??les distincts. Ces lacunes, ainsi que des donn??es exp??rimentales suppl??mentaires, ont donn?? lieu ?? l'hypoth??se ou "multi-composants" "variation de cofacteur".
En 2007, le biochimiste Surachai Supattapone et ses coll??gues Dartmouth College produite purifi?? prions infectieux de novo de composants d??finis (PrP C, lipides co-purifi??, et une mol??cule polyanionique synth??tique). Ces chercheurs ont ??galement montr?? que la mol??cule polyanionique n??cessaire ?? la formation de prion a ??t?? incorpor?? de mani??re s??lective dans des complexes de haute affinit?? avec les mol??cules de PrP, qui les conduit ?? ??mettre l'hypoth??se que les prions infectieux peuvent ??tre compos??s de plusieurs composants de l'h??te, y compris la PrP, un lipide, et des mol??cules polyanioniques, plut??t que PrP Sc seul.
En 2010, Jiyan Ma et ses coll??gues de l'Ohio State University produits prions infectieux d'une recette de la PrP exprim??e par une bact??rie recombinante, POPG phospholipides, et de l'ARN, confirmant l'hypoth??se multi-composant. Ce r??sultat est en contraste avec les ??tudes ont trouv?? que minimalement prions infectieux produits ?? partir de la PrP recombinante seule.
En 2012, Supattapone et coll??gues purifie la phosphatidyl??thanolamine lipidique de la membrane en tant que cofacteur endog??ne cellulaire capable de faciliter la formation de titre ??lev?? prions recombinantes d??riv??es ?? partir de plusieurs souches de prions. Ils ont ??galement signal?? que le cofacteur ??tait essentiel pour le maintien de la conformation infectieuse de la PrP Sc, et que les mol??cules de cofacteur dictent les propri??t??s de d??formation de prions infectieux.
Hypoth??se de l'empoisonnement par les m??taux lourds
Des rapports r??cents indiquent que le d??s??quilibre de l'hom??ostasie c??r??brale m??tallique est une cause importante de PrP Sc associ??e au g??ne neurotoxicit??, bien que les m??canismes sous-jacents sont difficiles ?? expliquer sur la base des informations existantes. Hypoth??ses propos??es comprennent un r??le fonctionnel de la PrP C dans le m??tabolisme de m??tal, et la perte de cette fonction en raison de l'agr??gation ?? la maladie associ??e forme PrP Sc comme la cause du d??s??quilibre cerveau m??tallique. Autres vues sugg??rent gain de fonction toxique par PrP Sc en raison de la s??questration de la PrP C associ??e au g??ne m??taux dans les agr??gats, r??sultant dans la g??n??ration de complexes de PrP Sc redox-actif. Les cons??quences physiologiques de certains PrP C interactions -m??tal sont connus, tandis que d'autres sont encore mal connues. Les cons??quences pathologiques de PrP C interaction -m??tal comprennent les dommages oxydatifs induits m??tallique, et dans certains cas, la conversion de la PrP C ?? une PrP Sc -comme forme.
Hypoth??se virale
L'hypoth??se de la prot??ine seule a ??t?? critiqu??e par ceux qui estiment que l'explication la plus simple des ??l??ments de preuve ?? ce jour est virale. Pour plus d'une d??cennie, Neuropathologiste Universit?? Yale Laura Manuelidis a ??t?? propose que les maladies ?? prions sont caus??es par un lieu non identifi?? virus lent. En Janvier 2007, elle et ses coll??gues publi?? un article faisant ??tat d'avoir trouv?? un virus dans 10%, ou moins, de leurs cellules infect??es par la tremblante dans la culture.
L'hypoth??se de virion indique que les EST sont provoqu??es par une mol??cule d'information ?? reproduire (ce qui est susceptible d'??tre un acide nucl??ique) li?? ?? la PrP. Beaucoup EST, y compris la tremblante et de l'ESB, montrent des souches ayant des propri??t??s biologiques sp??cifiques et distinctes, une caract??ristique qui les partisans de la sensation de hypoth??se de virion ne est pas expliqu??e par des prions.
Preuve en faveur d'une hypoth??se virale comprend:
- variation de souche: diff??rences dans l'infectiosit?? des prions, incubation, la symptomatologie et la progression parmi les esp??ces ressemble ?? celui observ?? entre les virus, en particulier les virus ?? ARN
- La longue p??riode d'incubation et l'apparition rapide des sympt??mes ressemble les lentivirus, tels que VIH induite par le SIDA
- Les particules virales comme qui ne semblent pas ??tre compos?? de PrP ont ??t?? trouv??s dans certaines des cellules de lign??es cellulaires ou la tremblante MCJ infect??es.
Des ??tudes r??centes se propageant infectiosit?? dans les r??actions sans cellules et dans les r??actions chimiques composant purifi?? sugg??rent fortement contre TSE nature virale. Plus r??cemment, en utilisant une recette d??finie similaire de plusieurs composants (PrP, POPG lipides, ARN), Jiyan Ma et ses coll??gues ont prions infectieux de la PrP recombinante exprim??e ?? partir E. coli, jetant plus de doute sur l'hypoth??se virale.
Champignons
Prot??ines fongiques pr??sentant changement conformationnel templated ont ??t?? d??couverts dans la levure Saccharomyces cerevisiae par Reed Wickner au d??but des ann??es 1990. Pour leur similitude m??caniste prions de mammif??res, ils ont ??t?? appel??s prions de levure. Par la suite, un prion a ??galement ??t?? trouv?? dans le champignon Anserina Podospora. Ces prions comportent de fa??on similaire ?? la PrP, mais sont g??n??ralement non toxique pour leurs h??tes. Le groupe de Susan Lindquist au Whitehead Institute a fait valoir certains des prions fongiques ne sont pas associ??s ?? un ??tat pathologique, mais peut avoir un r??le utile; Toutefois, les chercheurs du NIH ont ??galement fourni des arguments sugg??rant prions fongiques pourraient ??tre consid??r??s comme un ??tat maladif. Ainsi, la question de savoir si les prot??ines sont des maladies fongiques, ou ont ??volu?? pour certaines fonctions sp??cifiques, reste encore en suspens.
?? partir de 2012, il ya huit prot??ines prions connus dans les champignons, en sept Saccharomyces cerevisiae (Sup35, Rnq1, URE2, SWI1, MOT3, Cyc8 et Mod5) et une chez Podospora anserina (HET-s). L'article qui a rapport?? la d??couverte d'une forme de la prot??ine prion MCA1 a r??cemment ??t?? r??tract??e en raison du fait que les donn??es ne ont pas pu ??tre reproduits. En particulier, la plupart des prions fongiques sont bas??es sur les s??quences / glutamine ?? l'asparagine riche, ?? l'exception de HET-s et Mod5.
La recherche sur les prions fongiques a donn?? un fort soutien au concept de la prot??ine seule, puisque la prot??ine purifi??e extraite ?? partir de cellules avec un ??tat de prion a ??t?? d??montr?? pour convertir la forme normale de la prot??ine dans une forme mal repli??e in vitro, et dans le processus, de conserver les informations correspondant ?? diff??rentes souches de l'??tat de prion. Il a ??galement jeter une certaine lumi??re sur les domaines ?? prions, qui sont des r??gions dans une prot??ine qui favorisent la conversion en un prion. Prions fongiques ont permis de sugg??rer des m??canismes de conversion qui peuvent se appliquer ?? tous les prions, si prions fongiques apparaissent distincte de prions de mammif??res infectieuses en l'absence de cofacteur n??cessaire pour la propagation. Les domaines de prions typiques peuvent varier entre les esp??ces caract??ristiques, par exemple des domaines ?? prions fongiques ne sont pas trouv??s dans prions de mammif??res.
| Fongiques Prions | |||||
|---|---|---|---|---|---|
| Prot??ine | H??te naturel | Fonction normale | Etat Prion | Prion ph??notype | Ann??e identifi?? |
| Ure2p | Saccharomyces cerevisiae | Azote catabolique r??presseur | [URE3] | La croissance sur les sources d'azote pauvres | 1994 |
| Sup35p | S. cerevisiae | Traduction facteur de terminaison | [PSI +] | Augmentation des niveaux de suppression de b??tises | 1994 |
| HET-S | Podospora anserina | R??gule h??t??rocaryon incompatibilit?? | [Het-s] | Formation h??t??rocaryon entre les souches incompatibles | |
| Rnq1p | S. cerevisiae | Prot??ine facteur de mod??le | [RNQ +], [PIN +] | Favorise l'agr??gation d'autres prions | |
| MCA1 | S. cerevisiae | Putative de levure caspase | [MCA +] | Inconnu | 2008 |
| SWI1 | S. cerevisiae | Remodelage de la chromatine | [SWI +] | Faible croissance sur certaines sources de carbone | 2008 |
| Cyc8 | S. cerevisiae | R??presseur transcriptionnel | [+ OCT] | D??r??pression transcription de plusieurs g??nes | 2009 |
| MOT3 | S. cerevisiae | Facteur de transcription nucl??aire | [MOT3 +] | D??r??pression transcriptionnelle des g??nes ana??robies | 2009 |
| SFP1 | S. cerevisiae | Facteur de transcription putatif | [ISP +] | Antisuppression | 2010 |
Les traitements potentiels et le diagnostic
Les progr??s de la mod??lisation informatique ont permis aux scientifiques pour identifier des compos??s qui peuvent servir en tant que traitement pour des maladies caus??es par des prions, par exemple un compos?? qui se trouve ?? lier une cavit?? de la PrP C et stabiliser la conformation, ce qui r??duit la quantit?? de PrP Sc nuisible.
R??cemment, des anticorps antiprion capable de traverser la barri??re sang-cerveau-barri??re et le ciblage cytosolique de la prot??ine prion (un obstacle majeur en th??rapeutique contraire ?? prions) ont ??t?? d??crits.
Dans la derni??re d??cennie, des progr??s ont ??t?? signal??s traiter avec inactivation ultra-haute pression de l'infectiosit?? des prions dans la viande trait??e.
En 2011, il a ??t?? d??couvert que les prions pourraient ??tre d??grad??s par les lichens .
Il continue d'??tre un probl??me tr??s pratique avec diagnostic des maladies ?? prion, dont l'ESB et la MCJ. Ils ont une p??riode d'incubation de mois ?? des d??cennies au cours de laquelle il n'y a pas de sympt??mes, m??me si la voie de convertir la prot??ine PrP normale cerveau en la forme toxique li??e ?? la maladie PrP Sc a commenc??. ?? l'heure actuelle, il n'y a pratiquement aucun moyen de d??tecter de mani??re fiable PrP Sc, sauf en examinant le cerveau en utilisant des m??thodes neuropathologiques et immunohistochimiques apr??s la mort. L'accumulation de la forme PrP Sc anormalement repli??e de la prot??ine PrP est une caract??ristique de la maladie, mais elle est pr??sente ?? des niveaux tr??s faibles dans des fluides corporels facilement accessibles comme le sang ou l'urine. Les chercheurs ont essay?? de d??velopper des m??thodes pour mesurer la PrP Sc, mais il ya encore aucune m??thode enti??rement accept??es pour utilisation dans des mat??riaux tels que le sang.
En 2010, une ??quipe de New York d??crit la d??tection de PrP Sc m??me si initialement pr??sent ?? une seule partie dans une cent mille millions (10 -11) dans le tissu c??r??bral. La m??thode combine amplification avec une nouvelle technologie appel??e Surround fibre optique immunologique (SOFIA) et certains anticorps sp??cifiques contre la PrP Sc. Apr??s l'amplification, puis en concentrant tout PrP Sc, les ??chantillons sont marqu??s avec un colorant fluorescent en utilisant un anticorps de sp??cificit?? et finalement charg??es dans un tube de micro-capillaire. Ce tube est plac?? dans un appareil sp??cialement con??u de sorte qu'il soit totalement entour?? par des fibres optiques pour capturer toute la lumi??re ??mise une fois que le colorant est excit?? en utilisant un laser. La d??tection de la technique a permis de PrP Sc apr??s plusieurs cycles de conversion de moins que d'autres ont r??alis??, r??duisant sensiblement la possibilit?? d'artefacts, ainsi que l'acc??l??ration de l'essai. Les chercheurs ont ??galement test?? leur m??thode sur des ??chantillons de sang de moutons apparemment en bonne sant?? qui a continu?? ?? d??velopper la tremblante. Les cerveaux des animaux ont ??t?? analys??s une fois que tous les sympt??mes sont apparus. Les chercheurs pourraient donc comparer les r??sultats des tissus du cerveau et le sang pris une fois les animaux sympt??mes des maladies expos??es, avec du sang obtenu plus t??t dans la vie des animaux et des animaux non infect??s. Les r??sultats montrent tr??s clairement que la PrPSc pouvait ??tre d??tect??e dans le sang des animaux bien avant l'apparition des sympt??mes.