

r??action d'aldolisation
Contexte des ??coles Wikip??dia
Enfants SOS offrent un chargement complet de la s??lection pour les ??coles pour une utilisation sur les intranets des ??coles. Une bonne fa??on d'aider d'autres enfants est de parrainer un enfant
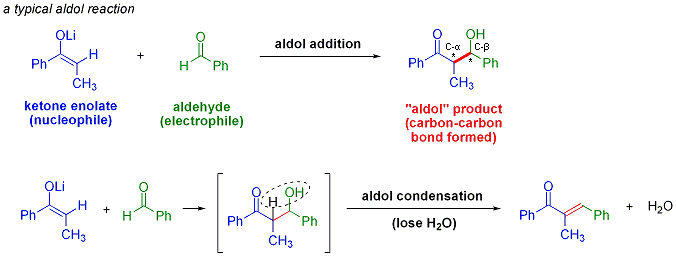
La r??action d'aldolisation est un important formation d'une liaison carbone-carbone r??action en chimie organique . Dans sa forme habituelle, il se agit de la addition nucl??ophile d'une c??tone ??nolate ?? une ald??hyde pour former une c??tone de β-hydroxy, ou "aldol" (ald ehyde + ALCOH ol) d'un motif structural trouve dans de nombreuses mol??cules naturelles et les produits pharmaceutiques. Parfois, le produit d'addition aldolique perd une mol??cule d'eau au cours de la r??action pour former un α, β-insatur??e. Ceci est appel?? un condensation d'aldol. La r??action d'aldol a ??t?? d??couvert de fa??on ind??pendante par Charles-Adolphe Wurtz et par Alexander Borodin Porfyrevich en 1872. Borodine observ?? la dim??risation aldolique de 3-hydroxybutanal partir l'ac??tald??hyde dans des conditions acides. La r??action d'aldolisation est largement utilis?? dans la production ?? grande ??chelle de produits chimiques de base tels que le penta??rythritol et le industrie pharmaceutique pour la synth??se de m??dicaments optiquement purs. Par exemple, route initiale de Pfizer pour le m??dicament Lipitor de maladie cardiaque (INN: atorvastatine), approuv?? en 1996, a employ?? deux r??actions aldoliques, permettant l'acc??s ?? des quantit??s multigramme ??chelle de la drogue.
Le motif structural aldolique est particuli??rement fr??quente chez polyc??tides, une classe de les produits naturels dont de nombreux produits pharmaceutiques sont d??riv??s, y compris le puissant immunosuppresseur FK506, la antibiotiques t??tracyclines, et l'agent antifongique amphot??ricine B. Des recherches approfondies sur la r??action aldolique a produit des m??thodes hautement efficaces qui permettent au contraire difficile synth??se de nombreux polyc??tides en laboratoire. Ceci est important car de nombreux polyc??tides, avec d'autres mol??cules biologiquement actives, se produisent naturellement en quantit??s impraticable petits pour compl??ment d'enqu??te. La synth??se de plusieurs de ces compos??s, une fois consid??r?? presque impossible, peut maintenant ??tre effectu??e en routine ?? l'??chelle du laboratoire, et se rapproche de la viabilit?? ??conomique ?? plus grande ??chelle, dans certains cas, tels que l'agent anti-tumoral tr??s actif discodermolide. En biochimie , la r??action d'aldolisation est une des principales ??tapes de glycolyse, o?? elle est catalys??e par des enzymes appel??es aldolases.
La r??action d'aldolisation est particuli??rement utile dans synth??se organique car il fabrique des produits avec deux nouvelles les centres st??r??og??nes (sur la α- et β-carbone du produit d'addition d'aldol, marqu?? d'un ast??risque dans le sch??ma ci-dessus). Les m??thodes modernes, d??crites ci-dessous, permettent maintenant la configuration relative et absolue de ces centres d'??tre contr??l??. Ceci est d'une importance particuli??re lors de la synth??se des produits pharmaceutiques, depuis mol??cules ayant la m??me st??r??ochimie connectivit?? de structure diff??rente, mais ont souvent des propri??t??s chimiques et biologiques tr??s diff??rentes.
Une vari??t?? de nucl??ophiles peut ??tre employ?? dans la r??action d'aldolisation, y compris la ??nols, ??nolates et ??nol ??thers des c??tones, des ald??hydes, et de nombreux autres compos??s carbonyl??s. Le partenaire ??lectrophile est un ald??hyde habituellement, bien que de nombreuses variantes, comme la R??action de Mannich, exister. Lorsque le nucl??ophile et ??lectrophile sont diff??rents (le cas habituel), la r??action est appel?? une r??action aldolique crois??e (par opposition ?? dim??res form??s dans une dim??risation aldolique).

Une solution de diisopropylamidure de lithium (LDA) dans le t??trahydrofuranne (THF) (dans le ballon ?? droite) est ajout?? ?? une solution de propionate de tert-butyle dans le ballon ?? gauche, formant son ??nolate de lithium. Un ald??hyde peut ensuite ??tre ajout?? pour initier une r??action d'addition aldolique.
Les deux flacons sont immerg??s dans une glace s??che / ac??tone bain de refroidissement (-78 ?? C) dont la temp??rature est surveill??e par un thermocouple (le fil sur la gauche).
M??canismes
La r??action d'aldolisation peut proc??der par deux m??canismes fondamentalement diff??rents. Les compos??s carbonyl??s tels que des ald??hydes et des c??tones, peuvent ??tre convertis en ??nols ou des ??thers d'??nols. Ces compos??s, ??tant nucl??ophile au α-carbone, peut attaquer carbonyles proton??s particuli??rement r??actifs tels que les ald??hydes proton??s. Ce est le "m??canisme d'??nol". Les compos??s carbonyl??s, ??tant des acides de carbone, peuvent ??galement ??tre d??proton?? pour former des ??nolates, qui sont beaucoup plus fortement nucl??ophiles que les ??nols ou des ??thers d'??nols et des ??lectrophiles peuvent attaquer directement. L'??lectrophile est un ald??hyde habituel, depuis les c??tones sont beaucoup moins r??actif. Ce est le ??m??canisme ??nolate".
Si les conditions sont particuli??rement dures (par exemple, NaOMe, MeOH, reflux), de la condensation peut se produire, mais cela peut g??n??ralement ??tre ??vit??s avec des r??actifs doux et des temp??ratures basses (par exemple, LDA (une base forte), le THF, -78 ?? C). Bien que l'addition aldolique proc??de g??n??ralement ?? proximit?? de r??alisation, la r??action ne est pas irr??versible, ??tant donn?? que le traitement de produits d'addition d'aldol avec les bases fortes induit habituellement r??tro-aldol clivage (donne les produits de d??part). condensations aldoliques sont irr??versibles.

M??canisme de Enol
Lorsqu'un catalyseur acide est utilis??, l'??tape initiale dans la M??canisme de r??action catalys??e par un acide comprend tautom??risation du compos?? carbonyle ?? l'??nol. L'acide sert aussi ?? activer le groupe carbonyle d'une autre mol??cule par protonation, rendant tr??s ??lectrophile. L'??nol est nucl??ophile ?? l'α-carbone, ce qui lui permet d'attaquer le compos?? carbonyle proton??e, conduisant ?? l'aldol apr??s d??protonation. Ce g??n??ral d??shydrate pour donner le compos?? carbonyle insatur??. Le sch??ma montre une auto-condensation catalys??e par un acide d'un ald??hyde typique.
Aldol catalys??e par un acide m??canisme
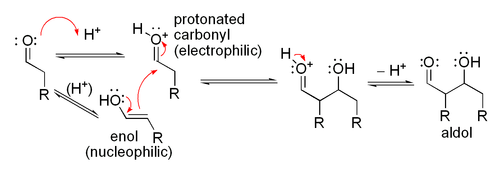
La d??shydratation catalys??e par un acide

M??canisme ??nolate
Si le catalyseur est une base mod??r??e telle que l'hydroxyde ou un ion un alcoxyde, la r??action d'aldolisation se fait par attaque nucl??ophile par la stabilis?? par r??sonance ??nolate sur le groupe carbonyle d'une autre mol??cule. Le produit est le sel alcoolate du produit d'aldolisation. L'aldol se est alors form??, et il peut ensuite subir une d??shydratation pour donner le compos?? carbonyle insatur??. Le sch??ma montre un m??canisme simple pour la base catalys??e aldol r??action d'un ald??hyde avec lui-m??me.
Base de r??action catalys??e aldolique (montr?? ?? l'aide - OCH 3 comme base)
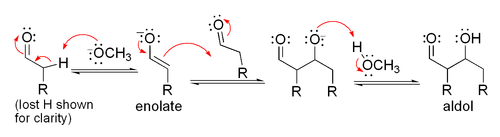
Base de d??shydratation catalys??e (parfois ??crit comme une seule ??tape)

Bien que seule une quantit?? catalytique de base est n??cessaire, dans certains cas, la proc??dure la plus courante consiste ?? utiliser un quantit?? stoechiom??trique d'une base forte telle que LDA ou NaHMDS. Dans ce cas, la formation de l'??nolate est irr??versible, et le produit d'aldolisation est pas form??e jusqu'?? ce que l'alcoxyde de m??tal du produit d'aldolisation est proton?? dans une ??tape de traitement final s??par??.
Zimmerman-Traxler mod??le
Des formes plus raffin??es du m??canisme sont connus. En 1957, Zimmerman et Traxler propos?? que certaines r??actions aldoliques ont ????tat de transition de six cha??nons s ayant une conformation chaise. "Ce est maintenant connu comme le mod??le Zimmerman-Traxler. E-??nolates donnent lieu ?? produits anti, tandis que Z-??nolates donnent lieu ?? produits syn. Les facteurs qui contr??lent la s??lectivit?? sont la pr??f??rence pour placer substituants ??quatoriale dans six cha??nons ??tats de transition et l'??vitement de interactions syn-pentane, respectivement. E et Z se r??f??rent ?? la relation st??r??ochimique cis-trans entre l'oxyg??ne ??nolate portant le contre-ion positif et le groupe de priorit?? la plus ??lev??e sur l'atome de carbone alpha. En r??alit??, seuls certains m??taux tels que le lithium et le bore suivre de mani??re fiable le mod??le Zimmerman-Traxler. Ainsi, dans certains cas, la r??sultat st??r??ochimique de la r??action peut ??tre impr??visible.

{kind=link}
Contr??le dans la r??action Aldol
Le probl??me
Le probl??me de ??contr??le?? dans le addition aldolique est le mieux d??montr?? par un exemple. Examiner les r??sultats de cette r??action hypoth??tique:
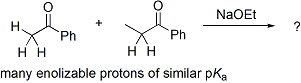
Dans cette r??action, deux c??tones non sym??triques sont condens??s ?? l'aide l'??thylate de sodium. La basicit?? de l'??thylate de sodium est telle qu'elle ne peut d??protoner totalement ou l'autre des c??tones, mais peut produire de petites quantit??s de l'??nolate de sodium de deux c??tones. En effet, cela signifie que en plus d'??tre ??lectrophiles potentiels d'aldolisation, les deux c??tones peuvent ??galement agir en tant que nucl??ophiles via leur ??nolate de sodium. Deux ??lectrophiles et nucl??ophiles deux puis des r??sultats potentiellement en quatre produits possibles:

Ainsi, si l'on souhaite obtenir un seul des produits crois??s, alors on doit l'addition aldolique "de contr??le".
Acidit??
Si un partenaire est beaucoup plus acide que l'autre, alors le contr??le peut ??tre automatique. Le proton plus acide est pr??lev??e par la base et un ??nolate est form??. Ce type de commande ne fonctionne que si la diff??rence de l'acidit?? est suffisamment grande et pas d'exc??s de base est utilis??e pour la r??action. Le contr??le est plus simple si seulement un des r??actifs a protons acides et seulement cette mol??cule constitue l'??nolate. Par exemple, l'addition de malonate de di??thyle dans benzald??hyde serait seulement produire un produit:
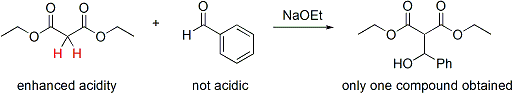
Dans ce cas, le doublement activ?? protons m??thyl??ne du malonate serait pr??f??rentiellement d??proton?? par l'??thoxyde de sodium et former quantitativement l'??nolate de sodium. Depuis benzald??hyde n'a pas de protons alpha-acides, il est seulement possible une combinaison nucl??ophile-??lectrophile; par cons??quent, le contr??le a ??t?? atteint. Notez que cette approche combine deux ??l??ments de contr??le: augmentation de l'acidit?? des protons alpha sur le nucl??ophile et l'absence de protons alpha sur l'??lectrophile.
L'ordre d'addition
Une solution commune est de former l'??nolate d'un partenaire de premier, puis ajouter l'autre partenaire dans contr??le cin??tique. Signifie que le contr??le cin??tique aldol r??action d'addition de l'avant doit ??tre nettement plus rapide que la r??action r??tro-aldol inverse. Pour que cette approche r??ussisse, deux autres conditions doivent aussi ??tre remplies; savoir, il doit ??tre possible de former quantitativement l'??nolate d'un partenaire et la r??action aldolique l'avant doit ??tre nettement plus rapide que le transfert de l'??nolate d'un partenaire ?? l'autre. Des conditions de contr??le cin??tique communes impliquent la formation de l'??nolate d'une c??tone avec LDA ?? -78 ?? C, suivie par l'addition lente d'un ald??hyde.
??nolates
Formation
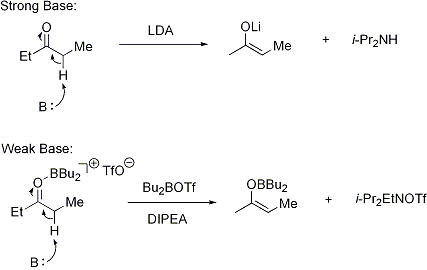
L'??nolate peut ??tre form??e en utilisant une base forte (??conditions difficiles??) ou en utilisant un Acide de Lewis et d'une base faible (??conditions douces??):
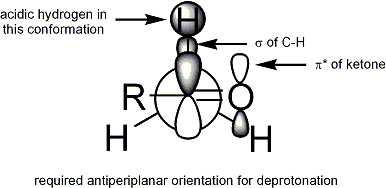
Pour d??protonation de se produire, la condition est que le st??r??o??lectronique alpha-CH liaison sigma doit ??tre capable de se chevaucher avec la pi * orbital de la carbonyle:
G??om??trie
Des ??tudes approfondies ont ??t?? r??alis??es sur la formation d'??nolates dans diverses conditions. Il est maintenant possible de produire, dans la plupart des cas, la g??om??trie de l'??nolate souhait??:

(- Dans l'image ci-dessus, le deuxi??me sch??ma de r??action devrait dire> 99% E-??nolate, pas Z -) Pour les c??tones, la plupart des conditions de ??nolisation donnent ??nolates Z. Pour esters, la plupart des conditions de ??nolisation donnent E ??nolates. L'addition de HMPA est connu pour inverser la st??r??os??lectivit?? de d??protonation.
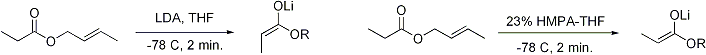
La formation st??r??os??lective des ??nolates a ??t?? rationalis??e avec le soi-disant mod??le Irlande, bien que sa validit?? est quelque peu discutable. Dans la plupart des cas, il ne est pas connue, le cas ??ch??ant, interm??diaires sont monom??re ou oligom??re dans la nature; n??anmoins, le mod??le Irlande reste un outil utile pour comprendre ??nolates.

Dans le mod??le Irlande, la d??protonation est suppos?? proc??der par un ??tat de transition monom??re six cha??nons. La plus grande des deux substituants sur l'??lectrophile (dans le cas ci-dessus, un groupe m??thyle est sup??rieure ?? protons) adopte une disposition ??quatoriale dans l'??tat de transition favoris??e, menant ?? une pr??f??rence pour E ??nolates. Le mod??le ne est manifestement pas dans de nombreux cas; par exemple, si le m??lange solvant est chang?? de THF ?? 23% HMPA-THF (comme on le voit ci-dessus), la g??om??trie de l'??nolate est inexplicable invers??e.
Vs Kinetic ??nolates thermodynamiques
Si une c??tone asym??trique est soumis ?? une base, il a le potentiel de former des ??nolates deux r??gioisom??res (en ignorant g??om??trie ??nolate). Par exemple:
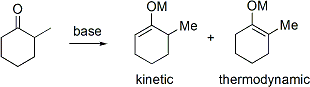
L'??nolate trisubstitu?? est consid??r?? comme le ??nolate cin??tique tandis que l'??nolate t??trasubstitu?? est consid??r?? comme l'??nolate thermodynamique. L'hydrog??ne alpha d??proton?? pour former l'??nolate cin??tique est moins encombr??, et donc d??proton?? plus rapidement. En g??n??ral, les ol??fines t??trasubstitu??es sont plus stables que des ol??fines trisubstitu??es en raison de la stabilisation hyperconjugaison. Le ratio de r??gioisom??res ??nolates est fortement influenc??e par le choix de la base. Pour l'exemple ci-dessus, contr??le cin??tique peut ??tre ??tablie avec LDA ?? -78 ?? C, donnant 99: une s??lectivit?? de cin??tique: ??nolate thermodynamique, tandis que le contr??le thermodynamique peut ??tre ??tablie avec triphenylmethyllithium au la temp??rature ambiante, donnant 10:90 s??lectivit??.
En g??n??ral, les ??nolates cin??tiques sont favoris??es par des temp??ratures relativement froides, des liaisons ioniques m??tal-oxyg??ne, et la d??protonation rapide en utilisant un l??ger exc??s d'une base forte, tandis que les ??nolates entrav?? thermodynamiques sont favoris??es par des temp??ratures plus ??lev??es, des liaisons covalentes relativement m??tal-oxyg??ne, et plus ??quilibration fois pour d??protonation en utilisant une petite quantit?? sous-stoechiom??trique d'une base forte. Utilisation d'une quantit?? sous-stoechiom??trique de base permet une petite fraction du compos?? carbonyle unenolized ?? ??quilibrer l'??nolate thermodynamique au r??gioisom??re en agissant comme une navette de protons.
St??r??os??lectivit??
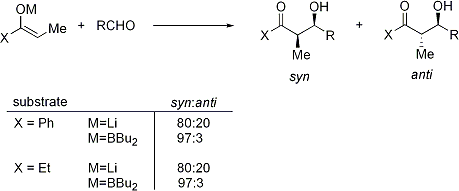
La r??action d'aldolisation est particuli??rement utile parce que deux nouveaux centres st??r??og??nes sont g??n??r??s dans une r??action. Des recherches approfondies ont ??t?? effectu??es pour comprendre le m??canisme de r??action et d'am??liorer la s??lectivit?? observ??e dans de nombreuses conditions diff??rentes. La convention de syn / anti est couramment utilis?? pour d??signer la st??r??ochimie relative ?? l'α- et β-carbone.

La convention se applique lorsque le propionate (ou ordre sup??rieur) nucl??ophiles sont ajout??s ?? ald??hydes. Le groupe R de la c??tone et le groupe R 'de l'ald??hyde sont align??s selon un motif "zig zag" dans le plan du papier, et la disposition des st??r??ocentres form??s est r??put?? syn ou anti, selon si elles sont sur le m??me ou c??t??s oppos??s de la cha??ne principale.
Documents plus anciens utilisent le ??rythro - thr??o nomenclature famili??re de la chimie des glucides.
E vs Z ??nolates
Il n'y a pas de diff??rence significative entre le niveau de stereoinduction observ?? avec E et Z ??nolates:


ion m??tallique
Le cation ??nolate de m??tal peut jouer un r??le important dans la d??termination du niveau de st??r??os??lectivit?? dans la r??action d'aldol. bore est souvent utilis?? parce que sa les longueurs de liaison sont sensiblement plus courte que celle des autres m??taux tels que le lithium , l'aluminium ou le magn??sium . Par exemple, les obligations de bore-carbone et de bore-oxyg??ne sont 1.4 ?? 1.5 ?? et 1.5 ?? 1.6 ?? de longueur, respectivement, alors que le m??tal-carbone et des liaisons m??tal-oxyg??ne typique sont g??n??ralement 1.9 ?? 2.2 ?? et 2,0 ?? 2,2 ?? de longueur, respectivement. Cela a pour effet de ??resserrement?? de la ??tat de transition:
St??r??os??lectivit??: Alpha st??r??ocentre sur le ??nolate
La r??action d'aldolisation peut pr??senter "st??r??ocontr??le ?? base de substrat", dans laquelle existant chiralit?? de chaque r??actif influe sur le r??sultat st??r??ochimique de la r??action. Cela a ??t?? largement ??tudi??, et dans de nombreux cas, on peut pr??dire le sens de induction asym??trique, si ce ne est le niveau absolu de diast??r??os??lectivit??. Si l'??nolate contient st??r??ocentre en position alpha, une excellente st??r??ochimique peut ??tre r??alis??e.

Dans le cas d'un ??nolate E, l'??l??ment de commande est dominant allylique 1,3 souche alors que dans le cas d'un ??nolate Z, l'??l??ment de commande dominante est d'??viter des interactions 1,3-diaxiales. Le mod??le g??n??ral est pr??sent?? ci-dessous:
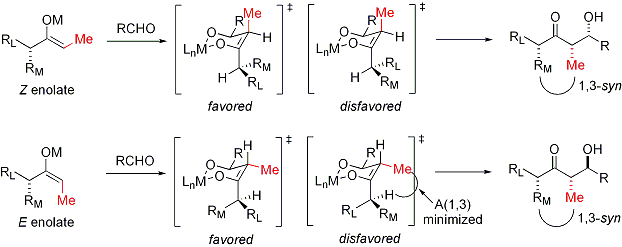
Pour plus de clart??, le st??r??ocentre sur l'??nolate a ??t?? ??pim??ris??; en r??alit??, le diastereoface oppos?? de l'ald??hyde aurait ??t?? attaqu??. Dans les deux cas, le diast??r??o-isom??re 1,3-syn est favoris??e. Il existe de nombreux exemples de ce type de st??r??ocontr??le:
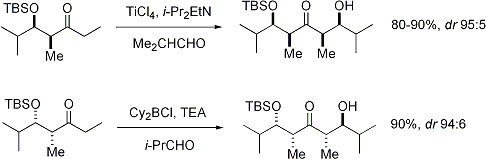
St??r??os??lectivit??: Alpha st??r??ocentre sur le ??lectrophile
Lorsque ??nolates attaque ald??hydes avec un st??r??ocentre alpha, excellente st??r??ochimique est ??galement possible. Le constat g??n??ral est que E ??nolates exposition Felkin s??lection de diastereoface, tandis que Z ??nolates pr??sentent anti-Felkin s??lectivit??. Le mod??le g??n??ral est pr??sent?? ci-dessous:

Depuis Z ??nolates doivent r??agir ?? travers un ??tat de transition qui contient soit une interaction syn-pentane d??stabiliser ou anti-Felkin rotam??re, Z-??nolates pr??sentent des niveaux inf??rieurs de diast??r??os??lectivit?? dans ce cas. Quelques exemples sont pr??sent??s ci-dessous:

St??r??os??lectivit??: mod??le fusionn?? pour stereoinduction
Si ?? la fois l'??nolate et la chiralit?? ald??hyde contiennent tous les deux pr??-existante, alors le r??sultat de la ??double stereodifferentiating" aldol r??action peut ??tre pr??dit en utilisant un mod??le st??r??ochimique fusionn?? qui prend en compte le biais ??nolate du visage, de la g??om??trie ??nolate, et ald??hyde biais du visage. Plusieurs exemples de l'application de ce mod??le sont pr??sent??es ci-dessous: 
Oxazolidinone la chimie de Evans
Synth??ses organiques modernes exigent maintenant la synth??se de compos??s dans forme ??nantiopur. Comme la r??action d'addition aldolique cr??e deux st??r??ocentres, jusqu'?? quatre st??r??oisom??res peuvent en r??sulter.

De nombreuses m??thodes qui contr??lent ?? la fois la st??r??ochimie relative (c.-??-syn ou anti, comme discut?? ci-dessus) et absolue st??r??ochimie (c.-??-R ou S) ont ??t?? d??velopp??s.
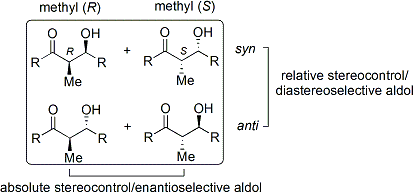
Une m??thode largement utilis??e est le Evans ' acyl Proc??d?? d'oxazolidinone. D??velopp?? dans les ann??es 1970 et 1980 par le David A. Evans et ses coll??gues, la m??thode fonctionne en cr??ant temporairement un ??nolate chiral en ajoutant un auxiliaire chiral. La chiralit?? pr??existante de l'auxiliaire est ensuite transf??r?? dans le produit d'addition aldolique en effectuant une r??action d'aldolisation diast??r??os??lective. Lors de l'enl??vement ult??rieur de l'auxiliaire, le st??r??oisom??re souhait?? d'aldol est r??v??l??.
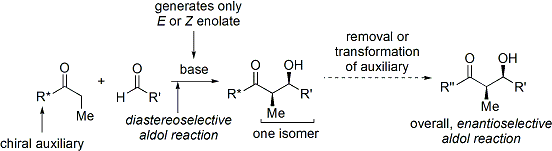
Dans le cas de la m??thode de Evans, l'auxiliaire chiral est un annex??e oxazolidinone, et le compos?? carbonyl?? est une r??sultante imide. Un certain nombre de oxazolidinones sont maintenant facilement disponibles dans les deux formes ??nantiom??res. Ceux-ci peuvent co??ter environ $ 10- $ 20 dollars par gramme, ce qui les rend relativement co??teux.
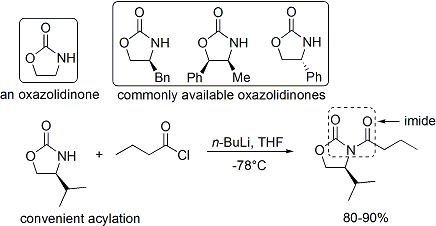
Le acylation d'une oxazolidinone est une proc??dure pratique et est officieusement appel?? ??chargement fait". Z-??nolates, conduisant ?? des adduits syn-aldoliques, peuvent ??tre form??s de mani??re fiable ?? l'aide de bore m??diation ??nolisation douce:

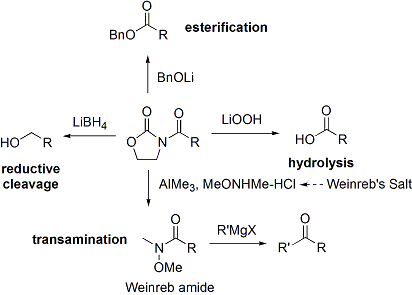
Souvent, un seul diast??r??o-isom??re peut ??tre obtenu par une cristallisation du produit d'addition aldolique. Malheureusement, les produits d'addition anti-aldol ne peuvent ??tre obtenues de mani??re fiable avec la m??thode Evans. Malgr?? le co??t et la limitation de ne donner que des produits d'addition syn, une fiabilit?? sup??rieure de la m??thode, la facilit?? d'utilisation et la polyvalence rendre la m??thode de choix dans de nombreuses situations. De nombreuses m??thodes sont disponibles pour le clivage de l'auxiliaire:
Lors de la construction imide, ?? la fois des r??actions d'addition aldolique syn et anti-s??lectives peuvent ??tre r??alis??es, ce qui permet l'assemblage de trois des quatre stereoarrays possibles: s??lective syn: et anti s??lective:
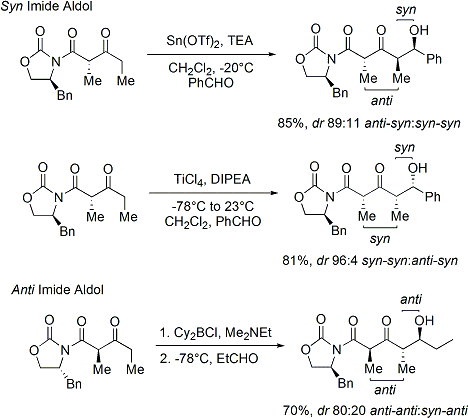
Dans les r??actions de syn-s??lective, les deux m??thodes donnent ??nolisation de l'??nolate de Z, comme pr??vu; cependant, le r??sultat st??r??ochimique de la r??action est contr??l??e par le st??r??ocentre de m??thyle, au lieu de la chiralit?? du oxazolidinone. Les m??thodes d??crites permettent l'assemblage st??r??os??lective polyc??tides, une classe de produits naturels qui pr??sentent souvent le r??tron aldolique.
Modern Chemistry Aldol
M??thodologie r??cente permet d??sormais une plus grande vari??t?? de r??actions d'aldolisation ?? mener, souvent avec une quantit?? catalytique de ligand chiral. Lorsque les r??actions utilisent de petites quantit??s de ??nantiom??riquement purs ligands pour induire la formation de produits ??nantiom??riquement purs, les r??actions sont g??n??ralement appel??s "catalytique asym??trique??; par exemple, un grand nombre catalytique diff??rent, aldoliques r??actions asym??triques sont maintenant disponibles.
Ac??tate de r??actions d'aldolisation
Une limitation cl?? de la approche auxiliaire chiral d??crit pr??c??demment est l'??chec de la N-ac??tyl imides de r??agir de mani??re s??lective. Une premi??re approche est d'utiliser un temporaire groupe thio??ther:
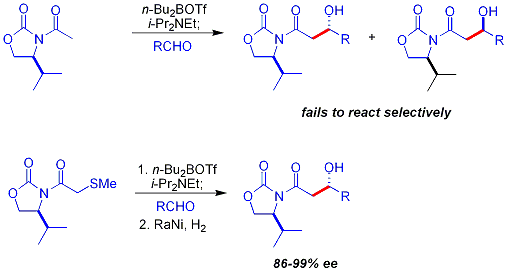
La r??action de Mukaiyama
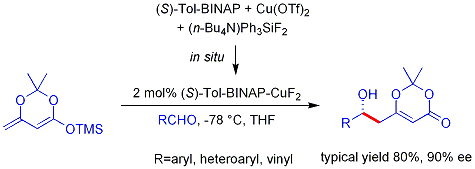
Le La r??action de Mukaiyama est le addition nucl??ophile de les ??thers de silyle ?? de ald??hydes catalys??e par un Acide de Lewis tel que le trifluorure de bore ou le chlorure de titane. La r??action d'aldol Mukaiyama ne suit pas le mod??le Zimmerman-Traxler. Carreira a d??crit la m??thodologie asym??trique particuli??rement utile avec des ac??tals de c??t??nes silyle, remarquable pour ses niveaux ??lev??s de ??nantios??lectivit?? et large champ de substrat.
La m??thode fonctionne sur ald??hydes aliphatiques non ramifi??s, qui sont souvent pauvres ??lectrophiles, pour proc??d??s catalytiques asym??triques. Cela peut ??tre d?? ?? la diff??renciation ??lectronique et st??rique pauvres entre leur enantiofaces.

L'analogue vinylogue processus de aldol Mukaiyama peut ??galement ??tre rendue catalytique et asym??trique. L'exemple ci-dessous fonctionne efficacement pour les ald??hydes aromatiques (mais pas aliphatiques) et le m??canisme est cens?? impliquer, un di??nolate de m??tal li?? chiral.
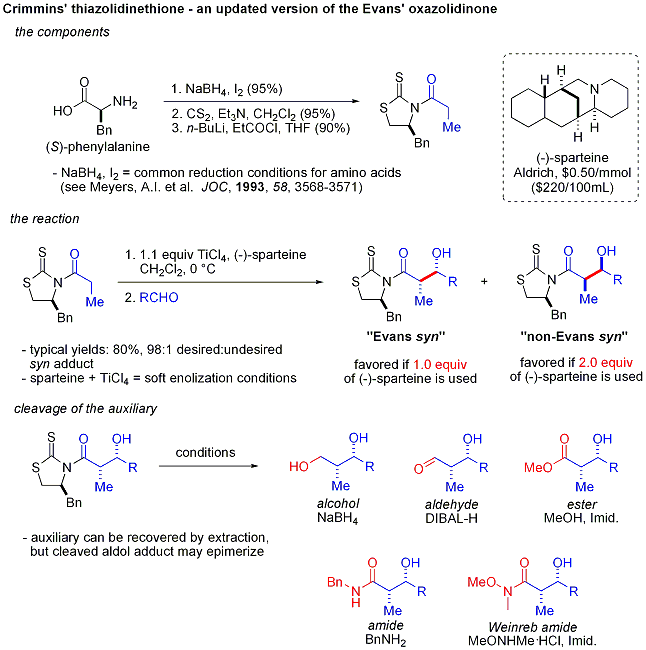
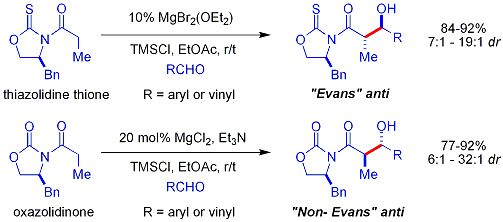
Crimmins thiazoldinethione aldol
Une version plus r??cente de l'auxiliaire du Evans est le thiazoldinethione Crimmins. Le rendements, diast??r??os??lectivit??s et ??nantios??lectivit??s de la r??action sont g??n??ralement ??lev??s, bien que pas aussi ??lev?? que dans les cas Evans comparables. Contrairement ?? l'auxiliaire Evans, cependant, le thiazoldinethione peut r??aliser des r??actions aldoliques ac??tate (ref:... Crimmins, Org Lett 2007, 9 (1), 149-152) et peut produire le ??syn Evans" ou "non-Evans syn" des produits d'addition en faisant simplement varier la quantit?? de (-) - Spart??ine. On pense que la r??action se d??roule par l'interm??diaire de six cha??nons, li?? au titane ??tats de transition, analogues aux ??tats de transition propos??es pour l'auxiliaire Evans.
R??actions aldoliques Organocatalytic
Un nouveau d??veloppement passionnant est l'utilisation de chiraux secondaires amine catalyseurs. Ces amines secondaires forment transitoire ??namines lorsqu'ils sont expos??s ?? des c??tones, qui peuvent r??agir avec des ??lectrophiles ??nantios??lectivement ald??hydes appropri??s. Ceci est connu comme la catalyse ??namine, un type de organocatalyse, puisque le catalyseur est enti??rement bas?? sur une petite mol??cule organique. Dans un exemple s??minal, proline efficacement catalys??e par la cyclisation d'un tric??tone:
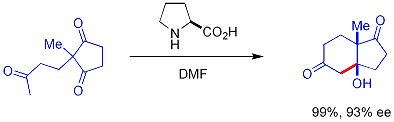
Cette r??action est connue comme la r??action Hajos-Parrish (aussi connu comme la r??action Hajos-Parrish-Eder-Sauer-Wiechert, se r??f??rant ?? un rapport de Schering contemporaine de la r??action dans des conditions plus s??v??res). Seule une quantit?? catalytique de proline est n??cessaire (3% en mole). Il n'y a pas de danger d'une r??action de base achiral parce que les interm??diaires ??namine transitoires sont beaucoup plus fortement nucl??ophiles que leurs ??nols de c??tones parent. Cette strat??gie est particuli??rement puissante car elle offre un moyen simple de g??n??rer ??nantios??lectivit?? dans les r??actions sans utiliser des m??taux de transition, qui ont les inconv??nients possibles d'??tre toxiques ou co??teux.
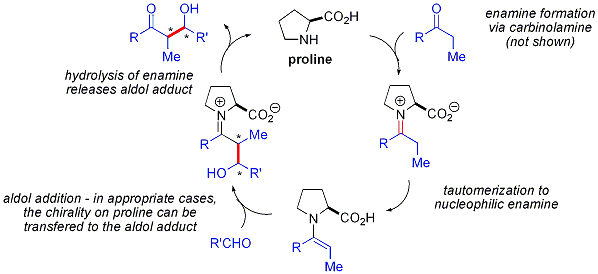
Fait int??ressant, les r??actions d'aldol catalys??e par la proline ne montrent pas d'effets non lin??aires (l'??nantios??lectivit?? des produits est directement proportionnelle ?? la ??nantiopuret?? du catalyseur). Combin?? avec preuves de marquage isotopique et des ??tudes informatiques , le projet de m??canisme de r??action pour les r??actions d'aldol catalys??e par la proline est la suivante:
Cette strat??gie permet ?? la r??action crois??e d'aldol autrement difficile entre deux ald??hydes. En g??n??ral, les r??actions crois??es entre aldoliques ald??hydes sont g??n??ralement difficiles parce qu'ils ne peuvent polym??riser facilement r??agir ou non s??lective pour obtenir un m??lange statistique des produits. Le premier exemple est pr??sent?? ci-dessous:
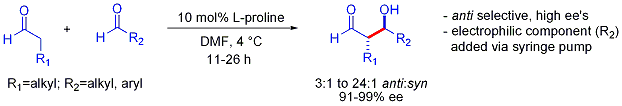
A la diff??rence de la pr??f??rence pour des produits d'addition syn typiquement observ??s dans des ajouts ?? base d'aldolisation-??nolates, ces ajouts d'aldolisation organocatalyzed sont anti-s??lectif. Dans de nombreux cas, les conditions organocatalytic sont suffisamment douces pour ??viter la polym??risation. Cependant, la s??lectivit?? n??cessite l'addition lente seringue-pompe command??e du partenaire ??lectrophile souhait?? car les deux partenaires ont g??n??ralement r??agissant protons ??nolisables. Si un ald??hyde n'a pas de protons ??nolisables ou alpha ou b??ta-ramification, un contr??le suppl??mentaire peut ??tre atteint.
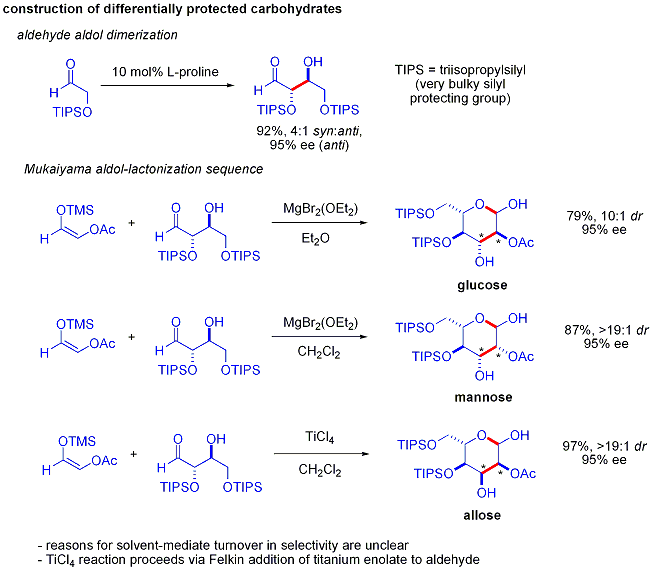
Une ??l??gante d??monstration de la puissance de r??actions aldoliques organocatalytic asym??triques a ??t?? divulgu??e par MacMillan et ses coll??gues en 2004 dans leur synth??se des diff??rentielle prot??g??es glucides . Bien que les m??thodes traditionnelles de synth??se accomplir la synth??se de hexoses aide de variantes du it??rative les strat??gies de protection-d??protection, ce qui n??cessite 8 ?? 14 ??tapes, organocatalyse peuvent acc??der ?? un grand nombre des m??mes substrats en utilisant un protocole efficace en deux ??tapes impliquant la dim??risation catalys??e par de la proline d'alpha-oxyaldehydes suivie d'une cyclisation aldolique Mukaiyama tandem.
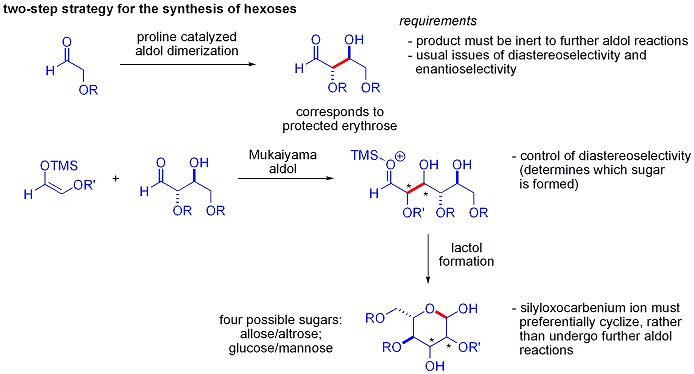
La dim??risation d'alpha-aldol oxyaldehydes exige que le produit d'addition d'aldol, un ald??hyde lui-m??me, soit inerte pour favoriser les r??actions d'aldolisation. Des ??tudes ant??rieures ont r??v??l?? que palier d'ald??hydes alpha-alkyloxy ou alpha- silyloxy substituants sont appropri?? pour cette r??action, tandis que des ald??hydes portant les groupes attracteurs d'??lectrons tels que ac??toxy ??taient non r??actif. La prot??g??e produit d'??rythrose pourrait ensuite ??tre converti en sucres quatre possibles par addition d'aldol suivie par Mukaiyama formation lactol. Cela n??cessite diastereocontrol appropri?? dans le addition aldolique Mukaiyama et le produit ions silyloxycarbenium ?? pr??f??rentiellement cyclisent, plut??t que de subir une autre r??action d'aldol. En fin de compte, le glucose , mannose, et allose ont ??t?? synth??tis??s:
additions "Direct" aldoliques
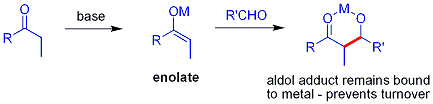
Dans l'addition aldolique d'habitude, un compos?? carbonyle est d??proton?? pour former l'??nolate. L'??nolate est ajout?? ?? un ald??hyde ou une c??tone, qui forme un alcoxyde, qui est ensuite proton?? sur bilan. Une m??thode sup??rieure, en principe, permettrait d'??viter la s??quence d??protonation-aldol-protonation en faveur d'une "addition aldolique directe". Le probl??me majeur dans un tel processus, ce est que l'addition aldolique g??n??re un alcoolate, qui est beaucoup plus fondamental que les mati??res premi??res, ce qui exclut le chiffre d'affaires de catalyseur:
Une approche, r??cemment d??montr?? par Evans, est de silyler le produit d'addition aldolique:
Cette m??thode est plus rentable et industriellement utile que les proc??dures fond??es ??nolates-plus typiques. Une approche plus r??cente, biomim??tique par Shair utilise b??ta-thioketoacids comme nucl??ophile. Le fragment est c??toacide d??carboxyl?? in situ (le est un ligand chiral bisoxazoline). Fait int??ressant, les ald??hydes aliphatiques et aromatiques ramifi??s sont g??n??ralement de mauvais substrats.
