
Maladie d'Alzheimer
Classification et ressources externes
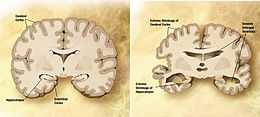
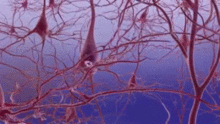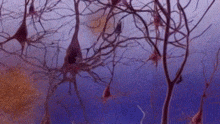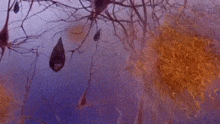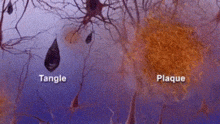
Comparaison d'un cerveau normal âgé (gauche) et du cerveau d'un patient atteint d'une maladie d'Alzheimer (droite).
Click on the following link to visit or download this HTML page
| CIM-10 | G30, F00 |
|---|---|
| CIM-9 | 331.0, 290.1 |
| OMIM | 104300 |
| DiseasesDB | 490 |
| MedlinePlus | 000760 |
| eMedicine | neuro/13 |
| MeSH | D000544 |
| GeneReviews | alzheimer |
La maladie d'Alzheimer (en allemand alʦhaɪ̯mɐ) est une maladie neurodégénérative (perte progressive de neurones) incurable du tissu cérébral qui entraîne la perte progressive et irréversible des fonctions mentales et notamment de la mémoire. C'est la forme la plus fréquente de démence chez l'être humain. Elle fut initialement décrite par le médecin allemand Alois Alzheimer en 1906[1].
Le premier symptôme est souvent des pertes de souvenirs (amnésie), se manifestant initialement par des distractions mineures, qui s'accentuent avec la progression de la maladie. Les souvenirs plus anciens sont cependant relativement préservés. L'atteinte neurologique s'étend par la suite aux cortex associatifs frontaux et temporo-pariétaux, se traduisant par des troubles cognitifs plus sévères (confusions, irritabilité, agressivité, troubles de l'humeur et des émotions, des fonctions exécutives et du langage) et la perte de la mémoire à long terme. La destruction des neurones se poursuit jusqu'à la perte des fonctions autonomes [réf. nécessaire] et la mort[2].
Deux types de lésions du cortex cérébral ont été mises en évidence dans la maladie d'Alzheimer : les plaques séniles et les dégénérescences neurofibrillaires. Cependant, les causes exactes de ces lésions restent encore inconnues. Il a néanmoins été montré que des facteurs génétiques et environnementaux contribueraient à l'apparition et au développement de cette maladie. Par ailleurs, des facteurs de risques ont été identifiés : certaines anomalies génétiques, des facteurs de risque cardio-vasculaires ou encore l'intoxication à certains métaux lourds.
Le diagnostic de la maladie d'Alzheimer repose essentiellement sur l'interrogatoire, des tests neuropsychologiques et sur la mise en évidence d'une atrophie corticale qui touche d'abord le lobe temporal interne et notamment l'hippocampe, régions importantes pour la mémoire. Elle est généralement diagnostiquée à partir de l'âge de 65 ans[3]. Des formes précoces, plus rares (moins de 5 % des patients), peuvent cependant apparaître beaucoup plus tôt. Les premiers signes de la maladie d'Alzheimer sont souvent confondus avec les aspects normaux de la sénescence, une dépression, un stress ou d'autres pathologies neurologiques comme la démence vasculaire. Elle fut ainsi sous-diagnostiquée jusque dans les années 1960.
La vitesse et l'évolution de la maladie sont variables d'un individu à l'autre, ce qui rend difficile tout pronostic précis. L'espérance de vie varie ainsi de 3 à 8 ans selon l'âge du patient au moment du diagnostic[4],[5]. Avec l'évolution de la maladie, les patients souffrent parfois de rejet de la part de la société et de leur famille[6],[7].
Il n'y a actuellement pas de traitement qui diminue la progression de cette maladie. Les soins proposés sont principalement d'ordre palliatif et n'ont qu'un effet limité sur les symptômes. La stimulation cognitive, l'exercice physique et un régime alimentaire équilibré pourraient retarder l'apparition de troubles cognitifs chez les personnes âgées[8]. Parce que la maladie d'Alzheimer ne peut être guérie et qu'elle est dégénérative, le patient s'appuie sur les autres pour l'aider. Le rôle de l'aidant principal est primordial[9],[10],[11],[12].
La maladie d'Alzheimer touchait environ 26 millions de personnes dans le monde en 2005 et pourrait en toucher quatre fois plus en 2050[13], ce qui équivaudrait alors à une personne sur 85[14]. Dans les pays développés, la maladie d'Alzheimer est l'une des pathologies les plus coûteuses pour la société[15],[16].
Étant donné la prévalence de la maladie, un important effort est mené par la recherche médicale pour trouver un traitement par médicament qui permettrait de stopper le processus neurodégénératif. La principale piste de recherche vise à s'attaquer aux plaques amyloïdes qui se forment entre les neurones au cours de la maladie et aux agrégats de protéines tau formant les dégénérescences neurofibrillaires à l'intérieur des neurones.
Stades d'évolution
La progression de la maladie d'Alzheimer est divisée en quatre stades, avec une progression caractéristique de troubles cognitifs.
Pré-démentiel
Les premiers symptômes sont souvent confondus avec les effets normaux du vieillissement ou du stress[6]. Des tests neuropsychologiques poussés peuvent cependant révéler des problèmes cognitifs légers jusqu'à huit ans avant que la personne ne remplisse les critères diagnostiques de la maladie d'Alzheimer[17]. Ces premiers symptômes affectent principalement les activités les plus complexes de la vie quotidienne[18]. Le déficit le plus notable est la perte de mémoire, qui se manifeste par des difficultés à se souvenir des faits récemment appris et à acquérir de nouvelles informations[17],[19]. Des problèmes plus subtils au niveau des fonctions exécutives comme l'attention, la planification, la flexibilité et l'abstraction ou encore des défauts de mémoire sémantique (mémoire du sens des mots et des concepts) sont également évocateurs des stades précoces de la maladie d'Alzheimer[17]. Une apathie peut être observée dès ce stade et reste le symptôme le plus persistant à travers l'évolution de la maladie[20].
Cette caractéristique préclinique de la maladie est également appelée trouble cognitif léger[19]. Cependant, le fait qu'il corresponde avec certitude au premier stade de la maladie d'Alzheimer reste controversé[21].
Léger
Chez les personnes souffrant de la maladie d'Alzheimer, l'évolution des symptômes (troubles de la mémoire, difficultés d'apprentissage, besoin d'aide dans l'accomplissement des tâches de la vie quotidienne) participe à la confirmation du diagnostic. Chez une partie de ces patients, des symptômes autres que les problèmes de mémoire apparaissent au premier plan et révèlent la maladie, notamment les problèmes liés au langage, aux fonctions exécutives, à l'identification (agnosie) ou encore à l’exécution des mouvements (apraxie)[22].
Les formes de mémoire à long terme
La maladie d'Alzheimer n'affecte pas de façon égale toutes les formes de mémoire. En effet, étant contrôlées par des structures cérébrales différentes, elles ne sont pas détériorées à la même vitesse par la maladie.
L'atteinte de la mémoire épisodique (création et gestion des souvenirs de la vie de la personne) est le trouble le plus précoce et le plus marqué dans la maladie d'Alzheimer, notamment avec des difficultés lors des étapes d'encodage, de stockage, de récupération des informations. Ces troubles peuvent être évalués avec le test de Gröber et Buschke[23].
La mémoire sémantique (les faits appris, comme « Rome est la capitale de l'Italie ») et la mémoire implicite (mémoire des gestes, comme faire du vélo) sont moins affectées au stade léger[24],[25].
Le langage
Les problèmes de langage (aphasie) sont caractérisés pour l'essentiel à ce stade par un « manque du mot » (ou aphasie léthologique), à l'origine d'un appauvrissement du vocabulaire et de la fluidité du discours ainsi que de l'expression orale et écrite[22],[26]. À ce stade, la personne touchée par la maladie d'Alzheimer est cependant toujours capable de communiquer des idées simples de manière adéquate[22],[26],[27].
La motricité
De même, bien que la personne reste capable de réaliser des tâches motrices fines, comme l'écriture, le dessin ou l'habillage, certaines difficultés apparaissent dans la coordination et la planification des mouvements (apraxie) mais sont rarement remarquées[22]. Au stade léger de la maladie, la personne reste indépendante lors des tâches courantes, mais va requérir de l'assistance ou de la supervision pour les activités complexes[22].
Modéré
La détérioration progressive des différentes fonctions cognitives conduit finalement à la perte d'indépendance : lorsque le sujet n'est plus capable de réaliser seul les activités les plus courantes[22]. Les difficultés du langage deviennent évidentes lorsque l'incapacité à se rappeler le vocabulaire (aphasie léthologique) conduit le patient à effectuer des substitutions incorrectes de mots (paraphasie) de plus en plus fréquentes. Les capacités de lecture et d'écriture se perdent progressivement[22],[27]. Avec la progression de la maladie, les séquences motrices complexes deviennent moins coordonnées, ce qui augmente les risques de chutes[22]. Durant ce stade, les problèmes de mémoire s'aggravent et la personne peut commencer à ne plus reconnaître ses proches[22]. La mémoire à long terme, jusque-là épargnée, commence à se détériorer[22].
Les changements comportementaux et neuropsychiatriques deviennent prévalents. Les manifestations classiques sont des errements, de l'irritabilité et une labilité émotionnelle qui conduit à des pleurs, des poussées d'agressivité soudaines ou de la résistance irrationnelle au soin[22]. Des périodes de grande confusion ont tendance à apparaître, notamment au coucher du soleil (la luminosité influant sur le caractère)[28]. Environ 30 % des patients Alzheimer développent des symptômes délirants et notamment des délires de changements d'identité[22]. Les patients perdent également la conscience de leur maladie et des limitations qu'elle entraîne (anosognosie)[22]. Enfin, ils peuvent souffrir d'incontinence urinaire[22]. Ces différents symptômes peuvent créer un stress important chez les proches et l'aide soignant, stress qui peut être réduit en passant d'un soin à domicile au placement en maison de soin spécialisée[22],[29].
Avancé
Durant la phase finale de la maladie d'Alzheimer, le patient est complètement dépendant du personnel de soin[22]. Le langage est réduit à quelques phrases simples ou même seulement des mots, ce qui conduit finalement à une perte complète de la parole[22],[27]. Malgré cette perte des capacités verbales, les personnes perçoivent encore les émotions de leur vis-à-vis et sont capables d'y répondre par des signes émotionnels[22]. Une certaine agressivité peut encore être présente, mais le plus souvent les conséquences de la maladie sont une extrême apathie couplée à un état de fatigue constant[22].
Les patients les plus avancés ne sont plus capables d'effectuer la moindre tâche motrice sans assistance[22]. La musculature et la mobilité sont détériorées au point que le patient reste alité et ne peut plus se nourrir seul[22]. La maladie d'Alzheimer est une maladie terminale, mais la cause de la mort est souvent due à un facteur externe, comme une infection des escarres ou une pneumonie, plutôt que la maladie elle-même[22].
Mécanismes pathologiques
Lors de la maladie d'Alzheimer, le cerveau du patient est victime d'un double processus de dégénérescence et d’inflammation. Il est caractérisé par deux types de lésions, chacune causée par une accumulation de protéines qui entraîne un dysfonctionnement de la cellule. Les progressions différentes de ces deux types de lésion participent à une lésion plus globale du cerveau :
- au niveau extracellulaire, l'accumulation du peptide β-amyloïde provoque des plaques amyloïdes ;
- au niveau intracellulaire, l'accumulation de protéine Tau s'appelle neurofibrilles.
Atrophie corticale
La maladie d'Alzheimer est caractérisée par une perte de neurones et de synapses dans le cortex cérébral et certaines régions subcorticales. Cette perte anormale entraine une atrophie des régions affectées, incluant le lobe temporal, pariétal et une partie du cortex frontal et du gyrus cingulaire[30]. Le cerveau peut ainsi perdre 8 à 10 % de son poids tous les dix ans, contre 2 % chez un sujet sain. L'atrophie corticale s'accompagne d'une dilatation des ventricules cérébraux et des sillons corticaux ainsi que d'une perte neuronale affectant particulièrement le système cholinergique (noyau basal de Meynert, septum, cortex entorhinal, amygdale et hippocampe).
Les études utilisant l'IRM et le PET scan ont documenté une réduction de certaines régions spécifiques chez les personnes atteintes de la maladie d'Alzheimer lorsqu'elles progressent d'un trouble cognitif léger vers une maladie d'Alzheimer, en comparaison des images de sujets sains âgés[31],[32].
Plaque amyloïde
_presenile_onset.jpg)
Origine
Les plaques amyloïdes correspondent à l'accumulation extracellulaire d’un peptide appelé « β-amyloïde » ou « peptide Aβ42 » (42, parce que constituée de 42 acides aminés). Cette protéine est une forme clivée anormale d'une glycoprotéine membranaire appelée « protéine précurseur de la protéine β-amyloïde » (ou APP pour Amyloïd Protein Precursor). C'est une enzyme, la β-secretase, qui provoque, pour des raisons encore mal comprises, le clivage anormal de la protéine APP. En temps normal cette protéine de la membrane des neurones est clivée par des secretases en peptide P3 non toxique.
Le peptide Aβ42 est un peptide insoluble qui ne peut être dégradé efficacement par les cellules environnantes. Il s'accumule dans le milieu extracellulaire, formant des plaques séniles qui compriment les neurones. Le peptide β-amyloïde est donc une protéine neurotoxique.
Mécanismes pathologiques
La présence de plaques amyloïdes entraîne un dysfonctionnement des neurones environnants, puis la mort neuronale par apoptose ou par nécrose.
Les plaques séniles libèrent de l'eau oxygénée, un peroxyde de formule H2O2, et entraînent un stress oxydant sur les neurones environnants. Dans H2O2, la liaison entre les deux atomes d'oxygène étant relativement faible, elle peut se cliver en présence d'un ion métallique ayant des propriétés redox (tels que le cuivre et le fer, tous deux présents dans le cerveau). Un radical OH° (hydroxyles), un des radicaux libres, est ainsi produit selon la réaction : H2O2 + Fe2+ = OH° + OH- + Fe3+ (réaction de Fenton). Les radicaux libres ne respectent pas la règle de l'octet. Ils sont donc instables car ils cherchent à coupler leur électron libre. Pour ce faire, ils vont arracher un atome d'hydrogène à la membrane du neurone (composée de molécules carbonées présentant de nombreux atomes d'hydrogène). La membrane « trouée » va donc laisser pénétrer d'autres radicaux libres qui s'attaqueront à l'ADN du neurone, entraînant la destruction des fonctions de la cellule privée d'information génétique. Dans le corps neuronal proprement dit, la membrane étant abîmée par les radicaux libres, les ions calcium et des fragments ß-amyloïdes vont également pénétrer et activer les phosphokinases à calcium (PKC) dont le rôle est d'éliminer la membrane neuronale abîmée. La PKC suractivée va éliminer des portions de membrane saines et accélérer le processus de destruction. Les radicaux libres et les fragments d'Aß42 vont pénétrer en surnombre dans le corps du neurone, affecter son fonctionnement et contribuer à l'apoptose.
D'autre part, le stress oxydatif provoque une réaction inflammatoire par le recrutement de la microglie qui va accélérer la destruction des neurones.
Autre hypothèse
Selon certains, il existerait une production cérébrale d'insuline. Cette production serait altérée dans le cadre de la maladie d'Alzheimer et pourrait être responsable des mécanismes en cause. Cette hypothèse est parfois dénommée diabète de type 3 par abus de langage, en référence au diabète sucré[33],[34].
Causes
L'apparition de plaque amyloïde peut être due au vieillissement normal. L'accumulation anormale sous-jacente à la maladie d'Alzheimer reste cependant inexpliquée. La responsabilité de toxiques est suspectée, tels que le mercure lorsqu'il est accumulé dans le cerveau sous sa forme ionisée divalente (Hg2+)- pro-oxydante et à forte affinité pour les groupements soufrés thiols[précision nécessaire].
Localisation et progression
Les plaques amyloïdes sont principalement localisées dans le néocortex et l'hippocampe
Dégénérescences neurofibrillaires

{kind=link}

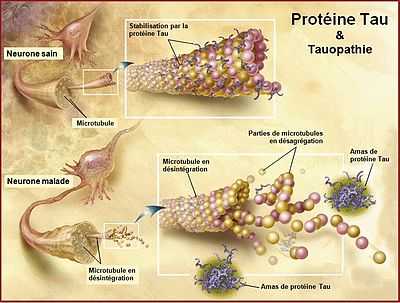
La protéine Tau est une macromolécule essentielle à la stabilité de la tubuline, protéine constituant majoritairement l'assemblage des microtubules qui forment le cytosquelette des axones. Les protéines tau se positionnent perpendiculairement à l'axone et assurent la rigidité des microtubules et le bon transport axonal.
Normalement, des protéines Tau se détachent périodiquement des microtubules, mais sont remplacées et rapidement dégradées chez le sujet sain. La maladie d'Alzheimer est caractérisée par des protéines Tau se détachant des microtubules et restant dans le milieu intracellulaire. Elles ne sont pas toutes dégradées et vont donc s'agréger. C'est cela qui va former les neurofibrilles. Trop de neurofibrilles bloquent le fonctionnement du neurone car elles ne permettent pas le transport axonal nécessaire à son activité. Les neurofibrilles compriment le neurone et provoquent une mort neuronale par apoptose.
Il existe plusieurs explications au détachement des protéines Tau, la principale reposant sur un problème de phosphorylation. La protéine tau est normalement peu phosphorylée. Lorsqu'elle est très phosphorylée, elle ne peut pas s'attacher aux microtubules. Dans la maladie d'Alzheimer, les protéines tau seraient hyperphosphorylées. Elles se détachent, se conforment en paire de filaments hélicoïdaux pathologiques et s'accumulent en formant des neurofibrilles. La cause de l'augmentation de la phosphorylation est inconnue. Une hypothèse avance que les radicaux libres, dus à la présence de plaques amyloïdes, détériorent la paroi membranaire des axones et laissent ainsi pénétrer des ions calcium qui vont sur-activer les Map kinases chargées de contrôler la phosphorylation des protéines tau. Ces protéines vont donc recevoir 5 à 9 groupements de phosphates, au lieu de 2 ou 3, et leur schéma de construction va changer.
Les coupures protéolytiques des protéines tau, qui sembleraient intervenir de façon précoce et seraient un événement concomitant à l'hyperphosphorylation de ces protéines.
Diversité génétique
Comme pour toutes les protéines, il existe un gène qui code la protéine Tau. Le gène présente notamment un motif appelé motif R qui permet à la protéine tau de se fixer sur les microtubules. Le gène tau a sept allèles différents qui peuvent être classés en deux catégories selon le nombre de motif R qu'ils possèdent :
- ceux à trois motifs R ;
- ceux à quatre motifs R.
Les protéines tau qui proviennent des allèles à trois motifs R ont une fixation moins importante que les protéines qui proviennent des allèles à quatre motifs.
Génétique
La majorité des cas de maladie d'Alzheimer sont dits sporadiques, ils ne sont pas héréditaires, bien que la présence de certains gènes soit un facteur de risque.
Cependant, environ 0,1 % des cas sont des formes génétiques familiales à transmission autosomale dominante (non-liée au sexe) qui se déclenchent habituellement vers 65 ans[37]. Cette forme de la maladie est connue comme la maladie d'Alzheimer familiale précoce (Early onset familial Alzheimer's disease).
La majorité de ces formes familiales autosomales dominantes peuvent être attribuées à une mutation sur un des trois gènes suivants[38] :
- le gène précurseur de la protéine amyloïde (APP), situé sur le chromosome 21 (cinq mutations du codon 717 sont connues) ;
- le gène Préséniline 1 (PSEN1) situé sur le chromosome 14 (principalement des mutations faux sens) ;
- le gène Préséniline 2 (PSEN2) situé sur le chromosome 1.
La plupart des mutations de ces trois gènes provoque une augmentation de la production de l'Aß42, le composant des plaques séniles amyloïdes[39]. Certaines mutations affectent uniquement le ratio entre la forme Aß42 et la forme normale Aß40, sans affecter le niveau de la protéine Aß42 elle-même[39],[40]. Cela suggère que les mutations de la preseniline peuvent causer la maladie même lorsqu'elle diminue la quantité totale d'Amyloïde-ß, ce qui suggère un rôle de cette protéine à un autre niveau du mécanisme pathologique que sur le seul fragment Aß (ex. : sur l'APP ou sur un autre fragment).
Facteurs de risque génétiques
Un des facteurs de risque génétiques les mieux décrits est la possession de l'allèle ε4 du gène de l'apolipoprotéine E (APOE)[41],[42]. Ce gène se présente sous trois allèles possibles chez l'homme : ε2, ε3 et ε4. Les allèles ε2 et ε3 sont spécifiques de l’espèce humaine. Ils proviennent d’une mutation du gène ε4. L’allèle le plus répandu est l’allèle ε3 (70 %), suivi de l’allèle ε4 (20 %) puis de l’allèle ε2 (10 %). Les allèles ε2 et ε3 sont associés au mécanisme de croissance neuritique (formation des axones et des dendrites). Cette croissance est importante pour la plasticité neuronale, un mécanisme assurant le bon fonctionnement et la longévité du système nerveux. Au contraire, l'allèle ε4 inhibe la croissance neuritique, diminue la plasticité neuronale et est par conséquent associé aux maladies du dysfonctionnement neuronal. Dans certaines populations, entre 40 et 80 % des personnes touchées par la maladie d'Alzheimer possèdent au moins une allèle APOEε4[42]. La présence de l'allèle APOEε4 augmente le risque de la maladie de trois fois à l'état hétérozygote et de quinze fois à l'état homozygote[37].
Le risque de maladie d'Alzheimer, toutefois, ne peut être expliqué uniquement par la génétique. Par exemple, chez certaines populations nigérianes, il n'y a aucune relation entre la présence de l'APOEε4 et l'incidence ou l'âge de déclenchement de la maladie d'Alzheimer[38],[43],[44].
Une mutation particulière du gène TREM2 augmente, dans à peu près les mêmes proportions que celle de l'APOE, le risque de contracter la maladie d'Alzheimer[45],[46]. La protéine codée par ce gène interviendrait dans l'élimination des neurones nécrosés par apoptose[47], la dysfonction de celle-ci pourrait alors contribuer à la survenue des lésions propres à l'Alzheimer[48].
Les généticiens s'accordent sur le fait que de nombreux autres gènes sont également des facteurs de risque ou, au contraire, ont des effets protecteurs et influent donc sur le déclenchement des formes tardives de l'Alzheimer. Néanmoins, des études comme celle effectuée au Nigeria et la pénétrance incomplète des facteurs de risque génétiques associés aux formes sporadiques indique une très forte influence de l'environnement. L'influence de plus de 400 gènes différents a été testée[38], la plupart sans donner de résultats[37].
- les personnes porteuses de trisomie 21 (syndrome de Down)[49],[50],[51].
- gène de la clusterine[52] ;
- gène codant la protéine PICALM (phosphatidylinositol-binding clathrin assembly protein) qui contribue au système immunitaire ;
- CR1 (récepteur 1 du composant 3b/4b du complément) a aussi été ajouté à la liste des gènes fortement suspectés[53].
Effets des lésions selon la localisation
Selon les endroits touchés par les lésions causées par la démence d'Alzheimer, les résultats seront différents :
- les lésions à la formation hippocampique occasionneront de l'amnésie ;
- les lésions au cortex frontal occasionneront des troubles d'attention et d'action ;
- les lésions au cortex postérieur occasionneront de l'aphasie, de l'agnosie et de l'apraxie ;
- les lésions au système limbique occasionneront des troubles émotifs ;
- les lésions au système réticulaire occasionneront des problèmes de vigilance et des troubles du sommeil.
Diagnostic
La maladie d'Alzheimer ne peut pas être diagnostiquée avec certitude. Seule l'autopsie, après le décès du patient, permet de confirmer avec certitude le diagnostic de maladie d'Alzheimer, grâce à un examen anatomo-pathologique du cerveau. Le recours à des biopsies de tissu cérébral est dangereux et s'avère peu utile. Le diagnostic de maladie d'Alzheimer est néanmoins donné lorsqu'un diagnostic clinique de démence est établi et que les examens complémentaires éliminent l'ensemble des autres diagnostics possibles (diagnostic par défaut)[réf. nécessaire].
Il faut noter d'autre part que des examens cérébraux post mortem peuvent attester d’un stade avancé de la maladie d'Alzheimer, sans que les malades n'aient présenté de signes cliniques de type altération cognitive. Ces observations ont entrainé le développement du concept de réserve cognitive (en), qui influe sur le profil d'évolution des signes cliniques : absence de signes au début de la maladie puis accélération rapide passé un « seuil » pour les personnes dotées d'une forte réserve[54].
Diagnostic clinique
Dépistage
Actuellement, il n'est pas recommandé de recourir au dépistage de la maladie, c'est-à-dire à la recherche de la maladie chez des personnes qui n'ont aucun symptôme[55]. En effet, en l'absence de traitement curatif, cette stratégie n'est pas pertinente au plan de la santé publique. Par contre, il est recommandé de reconnaître la maladie chez des personnes qui ont des symptômes ou des signes évocateurs. Il est alors question de détection et de diagnostic.
Signes précurseurs
On peut retrouver des signes précurseurs de la maladie d'Alzheimer jusqu'à 12 ans avant le diagnostic[56].
La maladie d'Alzheimer débute habituellement par des troubles de la mémoire. Certains patients remarquent que leur mémoire fonctionne moins bien qu'autrefois et consultent leur médecin pour cela. Chez d'autres patients, l'entourage plus que le patient lui-même remarque ses difficultés de mémoire. Les symptômes liés à la mémoire (plainte mnésique) ne sont cependant pas spécifiques de la maladie d'Alzheimer (voir diagnostics différentiels).
La maladie peut aussi se manifester par d'autres symptômes, comme une dépression, une perte d'indépendance fonctionnelle (nécessité d'une aide humaine dans les gestes de la vie quotidienne), des chutes répétées, un amaigrissement ou encore des troubles du comportement. À un stade plus évolué, d'autres troubles cognitifs apparaissent progressivement : altérations du langage, des gestes, de la motricité et de la communication. L'ensemble de ces symptômes ne sont pas spécifique d'un Alzheimer mais conduisent le patient à consulter un médecin qui le guidera le cas échéant vers un diagnostic en centre spécialisé.
Ce diagnostic est posé en deux étapes :
- rechercher l'existence d'un syndrome démentiel par un médecin pas forcément spécialisé ;
- rechercher la cause de ce syndrome démentiel.
Pour poser le diagnostic de démence, il est nécessaire de conduire une évaluation détaillée des fonctions cognitives qui est habituellement réalisée par un psychologue spécialisé en neuropsychologie.
Démence et maladie d'Alzheimer
Le terme démence signifie que les troubles cognitifs de la personne ont un retentissement dans son autonomie au quotidien[57]. La démence regroupe un ensemble de pathologies dont la maladie d'Alzheimer qui en est la forme la plus emblématique et la plus fréquente.
Critères de démence
Les critères de démence du DSM-IV reposent sur l'installation de troubles intellectuels portant de manière partielle ou complète sur :
- la mémoire : amnésie des faits récents puis anciens ;
- des troubles des fonctions exécutives : faire des projets, organiser, ordonner dans le temps, avoir une pensée abstraite ;
- une aphasie : perturbation du langage (manque du mot) ;
- une apraxie : altération de la capacité à réaliser une activité motrice malgré des fonctions motrices intactes (difficulté à s'habiller) ;
- une agnosie : impossibilité de reconnaître ou d’identifier des objets malgré des fonctions sensorielles intactes.
Ces troubles ont un retentissement socio-professionnel. Leur évolution se fait de manière progressive et irréversible (déclin continu, etc.). Enfin, ces signes ne peuvent être expliqués par d'autres causes : ni organique (tumorale, infectieuse, toxique), ni psychique (dépression, schizophrénie), et en dehors d'une confusion aiguë.
Outils d'évaluation cliniques
Le diagnostic de maladie d'Alzheimer nécessite l'évaluation de plusieurs fonctions cognitives. Certaines perturbations sont en faveur du diagnostic.
Tests de diagnostic précoce réalisables par un médecin
- Le MMSE (Mini Mental State Evaluation, ou Test de Folstein), est le test le plus répandu et celui recommandé par la Haute Autorité de santé[57]. Il comporte 18 questions ou tâches et il est réalisable en 15 minutes environ. Il fournit un score variant de 0 à 30 points : un score inférieur à 24 sur 30 est suspect de démence ; un score de 28 ou plus est normal. Ce résultat doit cependant être interprété selon le niveau éducatif du patient car un haut niveau peut améliorer le score et donc fausser le test (quel est son dernier diplôme obtenu, quel est son (ancienne) activité professionnelle). Il faudra s'assurer de l'absence de confusion avant sa réalisation. Il est recommandé de ne pas faire ce test dès l'arrivée des patients dans le services mais à distance de quelques jours [réf. nécessaire].
- Le test Codex[58] : ce test réalisable en 3 minutes combine une épreuve de mémoire et un test de l'horloge : ces éléments forment un arbre de décision complété pour certains patients par des questions sur l'orientation spatiale. Il est redondant avec certains autres tests (réalisés en routine en gériatrie hospitalière) mais il est plus rapide et particulièrement adapté à la médecine de ville[59].
- Le « test de l'horloge »[60] : il consiste à faire dessiner à la personne le cadran d'une montre. On demande au patient d'indiquer une heure choisie par l'examinateur. Par exemple, mettez une petite aiguille et une grande aiguille pour indiquer cinq heures moins le quart. Il est réalisable en 2 minutes environ.
- Le « test des 5 mots » de Dubois[61] : ce test évalue la mémoire en distinguant les processus d'encodage et de rappel.
- D'autres tests peuvent également être réalisés en pratique courante : le MoCA[62], les fluences verbales…
Outils d'évaluation cognitive en milieu spécialisé réalisés par un neuropsychologue
Outre les tests précédents, on peut citer :
- le test de Gröber et Buschke pour explorer la mémoire[63] ;
- le test Trail Making Test[64], pour explorer les fonctions exécutives et l'attention [réf. nécessaire] ;
- le test de Stroop pour explorer l'attention[65] ;
- les tests de dénomination, pour explorer le langage [réf. nécessaire] ;
- la copie de figures complexes (ex. : la figure complexe de Rey)[réf. nécessaire].
Examens complémentaires
Le diagnostic de démence est essentiellement clinique. Des examens complémentaires doivent être réalisés afin d'éliminer des causes curables.
Les examens biologiques recommandés sont[57] : dosage de la thyréostimuline hypophysaire (TSH), un hémogramme, une CRP, une natrémie, une calcémie, une glycémie, une albuminémie et un bilan rénal (créatinine et sa clairance). Les examens biologiques à réaliser en fonction du contexte sont : vitamine B12, folates, bilan hépatique (transaminases, gamma GT), sérologie syphilitique, VIH ou maladie de Lyme.
L'imagerie cérébrale est systématique :
- L'IRM est l'examen d'imagerie de choix pour le diagnostic étiologique des démences. Dans la maladie d'Alzheimer, elle peut montrer une atrophie corticale (on recherchera surtout une atrophie hippocampique en coupe coronale séquence T1), cependant l'atrophie corticale ou sous-corticale n'est pas spécifique de la maladie d'Alzheimer. Cet examen permet aussi d'éliminer notamment d'autres causes : tumeurs, accident vasculaire cérébral, hématome intra-cérébral ou sous-dural, encéphalopathie éthylique. Des indices sont cependant en cours d'évaluation pour tenter de faire un diagnostic précoce (dont la diminution de la taille de l'hippocampe). Cependant, cet examen est peu accessible, son coût est élevé et son délai d'attente est long.
- Le scanner cérébral est réalisé si l'IRM est contre-indiquée ou n'est pas réalisable. Son coût est faible et son délai d'attente est court. Il est moins précis que l'IRM, mais permet d'éliminer les causes curables sus-mentionnées.
D'autres examens sont réalisés dans certaines circonstances seulement :
- le dosage dans le liquide céphalo-rachidien de la protéine tau, des protéines tau phosphorylées et du peptide amyloïde bêta de 42 acides aminés sont réalisés après une ponction lombaire. Ces dosages ne sont pas réalisés en pratique courante car ils sont pratiqués lorsque le diagnostic étiologique de la démence est incertain. Ils sont réalisés dans certains centres hospitaliers spécialisés ;
- la tomographie à émission de positons ou TEP est un examen permettant l'analyse de certains traceurs radioactifs injectés dans l'organisme qui sont des marqueurs de la perfusion ou du métabolisme. On note une diminution assez nette du métabolisme de plusieurs parties du cerveau (lobe temporal, pariétal et postérieur) avec une bonne sensibilité et spécificité [réf. nécessaire]. La diminution de l'activité de l'hippocampe serait un indice prometteur. Cet examen n'est pas réalisé de façon courante. Il est utilisé lorsque le diagnostic étiologique de la démence est incertain, malgré l'évaluation clinique et l'IRM. La TEP peut aussi être utilisée avec des traceurs qui se lient aux plaques amyloides (PIB). Ce type d'examen est réalisé dans le cadre de protocoles de recherche ;
- la tomographie à émission monophotonique (TEMP) utilise aussi des marqueurs de perfusion ou du métabolisme cérébral. Cette imagerie fonctionnelle est utile pour le diagnostic étiologique des démences, lorsque les données cliniques ou d'imagerie IRM ne suffisent pas à poser le diagnostic. En particulier, la TEMP est utile pour identifier les démences fronto-temporales. La TEMP peut aussi être réalisée avec des traceurs du système dopaminergique : c'est le Dat-scan, un autre examen d'imagerie isotopique utile pour différencier la maladie d'Alzheimer de la démence à corps de Lewy, une autre démence dégénérative. Dans cette maladie, il y a une hypofixation du traceur au niveau du striatum (zone de fixation normale pour cet examen). La Dat-scan utilise un traceur fixant spécifiquement les transporteurs de la dopamine marquée à l'iode 123.
Diagnostics différentiels
On peut évoquer le diagnostic de maladie d'Alzheimer devant beaucoup de situations différentes. Néanmoins de nombreuses autres pathologies ou conditions peuvent expliquer certains symptômes et doivent être écartées avant d'établir de manière certaine le diagnostic de maladie d'Alzheimer.
- Au stade initial de la maladie, un oubli bénin, un déficit cognitif léger (mild cognitive impairment (MCI)).
- Troubles cognitifs liés aux médicaments (iatrogènes) : prise de benzodiazépines, médicaments à effet anticholinergique (antidépresseurs tricycliques, antihistaminiques de première génération, antispasmodiques anticholinergiques, neuroleptiques phénothiaziniques).
- Les troubles anxieux ou la dépression peuvent également entraîner des pertes de mémoire sans gravité. Cependant, ces troubles sont parfois associés à la maladie d'Alzheimer et un traitement d'épreuve par antidépresseur peut aider au diagnostic[réf. nécessaire].
- Le syndrome d'apnée du sommeil peut-être évoqué devant des ronflements, une somnolence et un surpoids. Un bilan spécialisé en centre du sommeil avec une prise en charge adaptée peut améliorer les troubles de la mémoire.
- Le syndrome confusionnel.
- Tout trouble d'origine métabolique (hypoglycémie, hyponatrémie, hypothyroïdie) ou toxique (drogues, alcool).
Une fois le diagnostic de démence posé, une évaluation cognitive globale, fonctionnelle, thymique et comportementale en centre mémoire spécialisé va permettre de poser le diagnostic étiologique de la démence[66] :
- Maladie d'Alzheimer
- D'autres formes de démences d'origine dégénératives comme la démence à corps de Lewy, les démences associées à la maladie de Parkinson, la démence frontotemporale (avec ses variantes : démence sémantique et aphasie primaire progressive), la chorée de Huntington, La paralysie supranucléaire progressive, la maladie de Creutzfeldt-Jakob…
- Démences non dégénératives comme la démence vasculaire, la démence traumatique, l'hydrocéphalie à pression normale, les démences métaboliques ou infectieuses…
- L'association d'un tableau évoquant la maladie d'Alzheimer avec des signes cliniques et/ou radiologiques de lésions cérébrales d'origine vasculaire est en faveur d'une démence mixte.
Facteurs de risque
Le premier facteur de risque reste avant tout l'âge (supérieur à 65 ans), ce qui fait de la maladie d'Alzheimer une maladie du vieillissement. Un seul facteur génétique a été retrouvé l'allèle ε4 du gène de l'apolipoprotéine E. Cependant, l'utilisation du génotypage dans la pratique courante ou le dépistage n'est pas recommandé à ce jour du fait de l'absence de prise en charge spécifique. Voir Génétique. La petite taille, surtout chez les hommes, semble corrélée à un risque plus élevé de contracter la maladie[67],[68].
Facteurs de risque cardiovasculaires
Les maladies cardiovasculaires relativement précoces (à partir du milieu de vie)[69],[70],[71],[72] peuvent être un facteur de risque.
Un traitement contre l'hypertension artérielle limite le risque de mourir précocement, mais aussi celui d'être admis pour une longue durée dans un centre de soins (risque diminué de 49 %), pour des raisons encore incomprises. De manière générale, l'hypertension est un facteur de risque de démence[73]. Certains se demandent même si la maladie d’Alzheimer n'est pas une maladie vasculaire plutôt que neurodégénérative[74], notamment liée à une hypoperfusion cérébrale[75] et à une mauvaise irrigation du cerveau[76], comme dans d'autres formes de démence peut-être[77].
L'hypercholestérolémie[78] est également un facteur de risque. Un régime riche en acides gras polyinsaturés oméga-3 et omega-6, et pauvre en acides gras saturés pourrait à l'inverse diminuer le risque de développer la maladie d'Alzheimer[79].
Le tabagisme augmenterait sensiblement le risque de survenue de la maladie d'Alzheimer[80].
Facteurs de risque médicamenteux
L'usage des anxiolytiques[81](en particulier des benzodiazépines[82]) et des somnifères[83] augmenterait le risque de survenance de la Maladie d'Alzheimer. Le risque serait majoré de 20 % à 50 %. La France est un pays de forte consommation de ces produits.
Facteurs de risques : maladies métaboliques
Le diabète est un facteur de risque. Les diabétiques courent un risque environ deux fois plus élevé d'être concerné par la démence vasculaire ou la maladie d'Alzheimer que le reste de la population[84]. Même pour les non-diabétiques un taux élevé de glucose élève significativement le risque[85].
Facteurs environnementaux autres
D'autres facteurs de risque de la maladie d'Alzheimer ont été évoqués[86] : des antécédents personnels de dépression[87], un niveau socio-culturel bas[88]. Une méta-analyse met en avant la fragilité de constitution comme facteur de risque[89]. Les hypothèses du traumatisme crânien[90],[91] ou l'exposition à l'aluminium[92],[93],[94] sont controversées. L'inactivité physique a été longuement étudiée et semble pouvoir être retenue comme facteur de risque[95].
Mise en cause du mercure
Certains indices suggèrent une relation causale entre l'exposition au mercure et la maladie d'Alzheimer[96]. En revanche, les amalgames dentaires contenant du mercure ont fait l'objet de nombreuses études qui n'apportent pas la preuve de leur responsabilité au regard des maladies neurodégénératives[97],[98].
Ce mercure aurait pour principale origine les plombages dentaires : l’OMS considère que le mercure-vapeur émis par les amalgames dentaires est la 1re source d’exposition mercurielle des populations occidentales[99]. Les amalgames perdent environ 50 % de leur mercure (soit 1/2 gramme environ par amalgame), en 10 ans, avant stabilisation, et de nombreuses études récentes ont confirmé que le taux de mercure du cerveau est corrélé au nombre d’amalgames[100],[101],[102],[103],[104].
Une revue[105] (des connaissances et études récentes, faite par J. Mutter montre une grande cohérence des études disponibles. Boyd Haley et ses collègues (Université du Kentucky) ont récemment montré comment le mercure induit une neurodégénérescence caractéristique de la maladie d’Alzheimer, à la suite d'une exposition chronique à de faibles doses de mercure-vapeur[106].
Une étude (de faible puissance statistique[réf. nécessaire] et portant sur des vétérans de l'US Army) avait en 1993 mis en évidence un taux 7 à 8 fois plus élevé de caries racinaires chez les victimes de maladie d'Alzheimer par rapport à un groupe témoin (apparié pour l’âge et le niveau d’éducation)[107]. Le mercure inhalé passe le mieux et plus directement au travers des poumons et qu'il est facilement stocké dans le cerveau qu'il gagne aisément grâce à sa lipophilie partielle. Ce sont les cellules gliales qui le stockent durablement, pour des dizaines d'années, sous forme d'un complexe insoluble soufré de cations mercuriques (Hg2+), après qu'il y a été oxydé par le peroxyde d’hydrogène dans une réaction catalysée par une catalase (la peroxydase).
En 2006, une étude suédoise montrait que les Suédois victimes de la maladie d'Alzheimer avaient plus souvent des problèmes dentaires que les non-malades[108].
Chez le rat de laboratoire exposé à du mercure, des processus de dégénérescence du cerveau sont observés, semblables à ceux de la maladie d'Alzheimer. Le cerveau de rats de laboratoire exposés à une dose de mercure-vapeur équivalente à celle de quelques amalgames dentaires, présente les mêmes anomalies moléculaires que celles observées à l'autopsie de 80 % des cerveaux de patients morts après une maladie d’Alzheimer. Et la gravité de l'anomalie est bien corrélée au taux de mercure mesuré dans le cerveau[109].
Un taux de mercure est mesuré plus élevé dans le cerveau des malades d'Alzheimer, et tout particulièrement dans le noyau basal de Meynert au centre de l'encéphale, là où la dégénérescence neuronale est la plus forte chez les malades[110],[111]. Les malades ont presque toujours un taux de mercure sanguin anormalement élevé (2 à 3 fois plus élevé que pour l'échantillon témoin)[112] et le taux sanguin du mercure est plus élevé chez les malades qui ont le plus de protéines β-amyloïdes se déposant dans le cerveau.
Le mercure et d'autres métaux lourds ont une affinité pour les groupements thiols, or la tubuline possède plusieurs cystéines, un acide aminé possédant une fonction soufrée thiol (indispensable à la polycondensation de cette protéine cytosolique et c'est cette polycondensation qui forme les microtubules rigides, principaux constituants du cytosquelette des neurones, nécessaire au transport axoplasmique qui nourrit le neurone. Les fonctions thiol ont une affinité très élevée pour les cations mercuriques hydrosolubles[113], elles interviennent d'ailleurs dans les phénomènes naturels de détoxication des métaux lourds.
In vitro, de faibles doses de mercure inorganique (Hg2+) suffisent à inhiber la production de glutathion par les neuroblastomes (cellules neuronales modifiées). Le glutathion est un tripeptide soufré normalement très présent dans le cerveau qu'il protège en tant que puissant antioxydant du milieu cellulaire. À faible dose, le mercure induit aussi une hyperphosphorylation des protéines tau et un dépôt de protéine β-amyloïde, deux caractéristiques de la maladie d’Alzheimer (démontré expérimentalement chez le porc)[114].
La mélatonine est une hormone essentielle, produite par la glande pinéale (ou épiphyse), synchronisant les biorythmes, mais jouant aussi un rôle antioxydant et protégeant les neurones du mercure. L'effet cytotoxique du mercure via le stress oxydatif est connu, mais des chercheurs ont montré dans les années 2000 qu'il augmente aussi la sécrétion de bêta-amyloïde et la phosphrylation des protéines Tau dans les cellules de neuroblastome SHSY5Y. Ils ajoutent que la mélatonine protège bien les neurones contre l’action oxydante des cations mercuriques[115]. In vitro, dans une culture de tissu cérébral humain, de très faibles doses de mercure inorganique suffisent à inhiber la phosphorylation de la tubuline par la guanosine-triphosphate (GTP), cofacteur nécessaire à la construction des microtubules, alors que d'autres métaux (même neurotoxiques connus comme le plomb, y compris sous leur forme ionisée) n'ont pas cet effet, pas plus que le fer ou le zinc ou l’aluminium, un temps suspectés[116].
Le mercure inhibe la fixation des riboses sur l’adénosine diphosphate (ADP ; coenzyme nucléotidique de la tubuline), ce qui inhibe la polycondensation de cette protéine (30), entraînant la formation d’amas neurofibrillaires cytotoxiques[117]. Des neurones mis en culture dégénèrent quand ils sont exposés à de très faibles doses de mercure inorganique ionisé (Hg2+) avec, dans le même temps, formation d’amas de neurofibrilles. L'Université de médecine de Calgary a produit une vidéomicroscopie qui montre qu'une dose infime de (10-7 Mol) mercure suffit à mettre à nu une gaine de microtubules, par perte des neurofibrilles qui se dépolymérisent progressivement (Voir la vidéo). Cet effet n’est pas retrouvé avec d’autres métaux neurotoxiques : aluminium, plomb, manganèse[118].
Des cultures de cellules souches de neurones ont été exposées à de faibles taux de mercure inorganique : celui-ci y bloque les fonctions de la tubuline, et induit la mort des cellules, par apoptose, ainsi que la formation de protéines connues pour être chaperonnes du stress thermique[119].
Des chélateurs (molécules captant les métaux) associés à des antioxydants permettent une solubilisation de la protéine β-amyloïde (PbA). C'est un indice de plus en faveur d'une responsabilité du mercure dans l'accumulation de cette protéine pathogène[120]. Un métabolisme anormal (homéostasie cellulaire perturbée) de deux autres cations métalliques (cuivre et zinc, nécessaires au fonctionnement du cerveau) serait également associé à l'apparition des plaques amyloïdes. Débarrasser le cerveau de ses excès de certains métaux pourrait être une piste thérapeutique contre la maladie d’Alzheimer[121]. In vitro, le mercure interagit négativement avec le glutamate (neuromédiateur excitateur) dont il perturbe la synthèse et le transport[122],[123]. Or, le glutamate est essentiel pour le cerveau et la mémoire, mais ne doit pas être présent en excès. Dans la fente synaptique, les cations mercuriques (à dose micromolaire, 10-6 Mol), freinent la capture du glutamate par les astrocytes, en se fixant sur les fonctions thiols des transporteurs protéiques du glutamate[124][réf. insuffisante]. Le glutamate s'accumule et devient hyperexcitant, au point de tuer les cellules nerveuses.
In vivo, le liquide cérébrospinal des M.A contient toujours un taux anormalement élevé de glutamine synthétase (GS), ce qui peut être dû au fait que le mercure inorganique freine l’activité de la GS[Quoi ?] dans les astrocytes bien plus efficacement que le cation méthylmercure (… avec une relation dose-dépendante, et même à très faible dose puisque 5 μM de mercure inorganique durant 6 heures suffisent à diminuer de 74 % l’activité de la GS)[125]. Le mercure, en augmentant le taux de glutamate excitotoxique peut entrainer la lyse d'astrocytes[126].
Le taux d'adénosine triphosphate (ATP) des cellules neuronales est en outre piloté par une enzyme (la créatine kinase, ou CK), qui est également vulnérable aux cations mercuriques car possédant un grand nombre de fonctions thiols. Or, un taux anormalement bas de cette enzyme est constatée dans les zones du cerveau les plus affectées par la maladie d’Alzheimer[127],[128].
Pronostic
Conséquences sociales
La famille comprend les enfants, les frères et sœurs, nièces et neveux, etc. En se référant au nombre estimé en 2007 de 800 000 malades en France et en considérant une moyenne de 3 cellules familiales autour d'un malade, ce sont plus de 2 400 000 personnes qui sont concernées plus ou moins directement par la maladie d'Alzheimer. C'est un problème majeur de société, la progression du nombre de malades étant d'environ 250 000 cas par an [réf. incomplète][129].
La famille a des ressources limitées en temps pour offrir à la personne malade le soutien dont elle a besoin de façon de plus en plus continue au fur et à mesure de l'évolution de la maladie. Pourtant, dans 70 % des cas, c'est la famille qui prend en charge la personne malade et lui permet de rester à domicile.
On a pris conscience de l'apport considérable de l'aidant naturel (aussi nommé aidant familial) et les professionnels se rendent compte que l'« aide aux aidants » est probablement une des manières de répondre à cet énorme défi de santé publique.
En France, 70 % des époux et 50 % des enfants d'une personne souffrant de la maladie d'Alzheimer lui consacrent plus de 6 heures par jour. 24 % des aidants – et 54 % s’il s’agit d’enfants d'une personne atteinte de la maladie d'Alzheimer – doivent réaménager leur activité professionnelle. 20 % des aidants naturels d'une personne atteinte de la maladie d'Alzheimer déclarent différer, voire renoncer à une consultation, une hospitalisation ou un soin pour eux-mêmes par manque de temps[130][réf. incomplète]. Si la prévalence de la maladie continue d'augmenter, elle mobilisera à elle seule et pour le simple nursing un dixième de la population active. Par ailleurs, il semblerait que la mortalité des personnes aidant soit supérieure à celle de personnes du même âge ne s'occupant pas d'un malade, mais cela n'est pas clairement établi à ce jour[131]. Dans le cadre du Plan Alzheimer 2008-2012, a été mis en place un métier le Technicien-Coordinateur de l’aide psycho-sociale aux aidants en réponse aux problématiques de ces aidants.
Traitements
Traitement préventif
Aucune méthode ne protège définitivement de la maladie d’Alzheimer, mais des facteurs de diminution du risque sont connus :
Activité cognitive et niveau scolaire
Une activité cognitive aide à réduire les risques de la maladie, la dégradation des facultés intellectuelles étant d'autant réduite que le nombre d'activités (tricot, TV, cinéma, lecture, jeux…) augmente[132].
La durée des études avant 25 ans n'a pas d'effet sur les pathologies à l'origine de la maladie d’Alzheimer, mais il protège des symptômes : le cerveau peut être atteint, mais les signes cliniques de dégénérescence cognitives sont retardés de 7 à 10 ans. Ce retard aboutit à diviser par deux la possibilité d'en manifester les symptômes pour la population la plus instruite[133]. Cet effet retardateur des symptômes a été constaté dans de nombreuses études, bien qu'elles présentent toutes des biais et aboutissent à des résultats détaillés divergents. Les différences portent sur l'effet protecteur des études dans le cas du stade de la démence grave, ou dans le cas de cerveaux de poids élevé auquel cas l'effet protecteur est divergent. Les mécanismes mis en œuvre dans cette faculté à compenser les causes de la maladie, désignés comme « réserve cognitive » ne sont pas connus, et il n'y pas de certitudes sur l'effet ou non d'autres facteurs tels que le style de vie dans les périodes ultérieures de la vie[134].
Activité physique
L'exercice physique tout au long de la vie pourrait prévenir le risque de maladie d'Alzheimer chez les sujets à risque[135], peut-être en diminuant le risque d'hypertension et d'accident cardiovasculaire[136].
Alimentation équilibrée
- pauvre en cholestérol[137],[138],[139]. Le cholestérol semble avoir un effet négatif sur les processus de mémorisation et sur certaines structures du cerveau;
- une alimentation moins riche en viande : plusieurs études cliniques tendent à démontrer que la consommation de viande[140](y compris les œufs) en grande quantité favoriserait le développement de la maladie d'Alzheimer[141],[142],[143],[144],[145]. Outre l'augmentation du risque liée au cholestérol, il est notamment évoqué dans certaines études le rôle de la méthionine, transformé en homocystéine par le métabolisme intermédiaire. Une hyperhomocystéinémie est un facteur augmentant le risque cardiovasculaire, et semblerait jouer un rôle dans l'apparition de la maladie d'Alzheimer. Cependant, il est nécessaire de rappeler que la méthionine est un acide aminé tout à fait indispensable à la vie, qui lorsqu'il est consommé dans les quantités habituelles ne pose aucun problème de santé publique.[réf. nécessaire] Selon certaines théories, la hausse du nombre de cas développant la maladie d'Alzheimer pourrait correspondre à la hausse de la consommation de viande dans le monde[142] : ainsi, une recherche de l’American Society for Nutrition (en), concernant des populations d'Amérique latine, de Chine et d'Inde, conclut que « la consommation de viande a été plus élevée chez ceux dont on a diagnostiqué une démence[146]. ». La consommation de poisson semble avoir un effet protecteur selon certaines études[147], ou pas d'effet[148], mais le poisson contient également de la méthionine. Les études sont intéressantes quand elles comparent deux groupes de même origine ethnique et du même âge qui ne consomment pas les mêmes aliments: japonais émigrés à Hawaii[149] et africains envoyés aux États-Unis[150]. À chaque fois l'alimentation riche en viande et graisses saturées est liée à un taux plus élevé de maladie d'Alzheimer. Cette recommandation concerne à la fois les protéines animales et les matières grasses d'origine animale[151] ;
- riche en vitamines C et E provenant de l'alimentation[152] ;
- pauvre en matières grasses[153],[151]. Les matières grasses consommées devant préférentiellement être polyinsaturées en évitant les matières grasses saturées et les acides gras trans[154] ;
- pauvre en sel (pour limiter le risque d'hypertension[155]) ;
- une limitation de l'apport en sucre. Les produits de dégradation du fructose (AGE pour Advanced Glycation End-Products) pourraient favoriser le vieillissement[156]. Certaines études posent le postulat que la maladie d'Alzheimer est une sorte de diabète de type 3[157]
- une limitation de l'apport en aliments à haut indice glycémique, ceux-ci ayant une influence néfaste sur la mémorisation[158]
- parallèlement, une alimentation végétarienne aurait un effet protecteur[159],[153]. Les populations indiennes pratiquant le végétarisme (depuis des générations) ont un taux d'individus touchés par la maladie d'Alzheimer qui est le plus faible enregistré de par le monde[160],[161] ;
- le régime méditerranéen aurait également un effet protecteur[162].
- le régime alimentaire traditionnel japonais (riz, légumes, poisson, pauvre en viande, sucre). La prévalence est 10 fois moindre au Japon qu'en France, mais progresse parallèlement à l'adoption d'un régime plus proche de celui des autres pays développés[163]
- riche en oméga-3. L'effet préventif semble probable[164], mais pas l'effet curatif[165] ;
- café et caféine ont été proposés comme l'un des moyens d'améliorer l'état des patients[166],[167], mais avec des effets secondaires possibles ;
- « Le thé vert aurait une incidence directe sur les fonctions cérébrales, car il contribue à la préservation de ces fonctions et à la réparation des cellules endommagées[168] » et réduirait le risque d'être atteint de démence et d'autres maladies neuro-dégénératives comme le Parkinson et l'Alzheimer[169] ;
- certains nutriments méditerranéens typiques semblent avoir un effet protecteur plus marqué, en particulier l'huile d'olive[170].
- d'autres compléments alimentaires sont étudiés actuellement[171],[172], des antioxydants d'origine naturelle en général, que l'on trouve dans certains fruits et légumes: carotéinoïdes dont l'astaxanthine et le lycopène, polyphénols dont les acides phénoliques présents dans le thé, les flavonoïdes, les stilbènes - dont le resvératrol contenu dans le raisin - et les lignanes. Aussi étudiés, les proanthocyanides, qui sont des polyphénols présents dans certains fruits rouges, ainsi que la curcumine[173] contenue dans le curcumin, un composant du curry.
Des recommandations alimentaires combinant régime méditerranéen et d'autres facteurs évoqués ci-dessus ont été développées spécifiquement pour réduire le risque de maladie d'Alzheimer[174], suivre ces recommandations permettrait de réduire le risque de 53%[175].
Un traitement expérimental combinant tous les facteurs alimentaires à éviter et les facteurs protecteurs, ainsi qu'une activité physique élevée, semble avoir une certaine efficacité[176].
Jeûne et restriction calorique
Plusieurs études indiquent que le jeûne, la restriction calorique ou le jeûne intermittent auraient un effet protecteur contre les maladies dégénératives du cerveau[177],[178],[179].
Lumière et rythmes biologiques
- L'exposition à la lumière naturelle semble améliorer certains symptômes. La prise de mélatonine, associée à une luminothérapie pourrait améliorer les troubles du sommeil, en agissant comme inducteur de sommeil mais aussi comme facteur d'allongement de la durée de celui-ci[180]. L'exposition à la lumière naturelle diminuerait aussi chez ces malades les symptômes de dépression (-19 %), les limitations fonctionnelles au quotidien (- 53 %), la détérioration cognitive (- 5 %). Chez les malades observés, la prise de mélatonine facilitait l'endormissement. L'association lumière + mélatonine a aussi diminué les comportements agressifs (- 9 %), les phases d'agitation et de réveil nocturne. Il est donc conseillé[181] « de bien éclairer les pièces en journée (… et) à l'inverse, de diminuer les sources de lumière en soirée pour que l'organisme reçoive le signal que la nuit est là ».
Traitements médicamenteux des facteurs de risques cardiovasculaires
Les traitements médicamenteux contre les facteurs de risques cardio-vasculaires semblent diminuer la survenance de la maladie d'Alzheimer, ou la reculer.
- Les traitements contre l'hypertension[182],[183],[184], et notamment les diurétiques, et surtout ceux qui agissent sur le potassium, ont été associés à un moindre risque de maladie d’Alzheimer[185]. Ils diminuent le risque de n'importe quelle forme de démence.
Les antihypertenseurs visant l'angiotensine semblent également diminuer le risque de MA[186].
- traitement contre l'hypercholestérolémie par des statines[187].
- la prise régulière d'aspirine ou d'anti-inflammatoires non stéroïdiens pourrait réduire le risque[188],[189].
Prise en charge dès les premiers signes
En France, un patient atteint de la maladie d'Alzheimer sur deux n'est pas diagnostiqué, et donc sans prise en charge adaptée[190]. Un dépistage et une prise en charge précoce sont conseillés pour favoriser le maintien à domicile le plus longtemps possible[191].
Approche non-médicamenteuse
L'approche non-médicamenteuse est une dimension importante de la prise en charge. Des approches très diverses sont proposées. Peu ont une efficacité réellement documentée par des études scientifiques. Elles ont l'avantage de ne pas avoir d'effets indésirables.
- La stimulation cognitive permettant de stabiliser les troubles cognitifs des personnes atteintes de la maladie d'Alzheimer, des équipes spécialisée Alzheimer à domicile (ESAD ont été mises en place suite à la mesure 6 du plan Alzheimer 2008-2012. Ces équipes proposent de la stimulation cognitive, mais également des mises en situation pour travailler les activités de la vie quotidienne et permettre à la personne de rester autonome à son domicile plus longtemps. Les interventions envers les aidants familiaux des patients semblent capables de retarder l'entrée en institution gériatrique, en particulier les interventions d'un type éducatif.
- Les aidants familiaux, par leur manière de se comporter, peuvent contrôler les troubles psychocomportementaux des malades. L'éducation des aidants, la notion de « base de sécurité » (un aidant choisi par le malade pour se sécuriser), de réseau d'aidants (autour de l'aidant principal), de tuteur de résilience pour l'aidant, sont autant d'éléments qui font de l'aidant familial un « traitement » en soi[192].
Prises en charge sociale
Le maintien à domicile le plus longtemps possible est souvent la solution demandée par les patients mais elle n'est pas toujours possible. La prise en charge sociale consiste à trouver une solution pérenne la plus adaptée à la personne en fonction de ses souhaits et de ses capacités. Les démarches sont souvent faites par les familles aidées des médecins et des assistantes sociales.
Traitements médicamenteux
Il n'existe actuellement aucun traitement pour guérir, ni même arrêter l'évolution, de la maladie d'Alzheimer.
Médicaments déconseillés chez le patient atteint d'une Maladie d'Alzheimer
Tous les médicaments ayant une action sur le cerveau ont un risque d'aggraver les troubles cognitifs des personnes atteintes d'une démence. Les principales classes thérapeutiques qu'il convient d'éviter[193] sont : les médicaments à effet anticholinergique et les psychotropes.
Utilisation des psychotropes chez le patient atteint de la maladie d'Alzheimer
Peu d'études ont été réalisées chez le patient atteint de la maladie d'Alzheimer concernant l'utilisation des psychotropes[194]. La plupart des recommandations sont faites à partir d'extrapolation des données issues des patients jeunes ou de l'expérience clinique.
Les études réalisées chez les patients atteints d'une maladie d'Alzheimer montrent que ces médicaments sont malgré tout très utilisés[195] : ils sont prescrits chez plus de 2/3 des patients atteints d'une maladie d'Alzheimer.
- Les antidépresseurs : la prévalence de la dépression est estimée entre 37 % et 50 % des patients[196],[197]. Ils n'ont pas prouvé d'efficacité[198] sur l'évolution de la maladie d'Alzheimer, mais ils peuvent avoir une efficacité sur la dépression associée, l'anxiété et les troubles du comportement[199].
- Les neuroleptiques sont fortement déconseillés chez les patients atteints d'une maladie d'Alzheimer[194],[200] sauf dans des situations particulières d'agitation majeure avec des risques de blessure pour le patient ou les soignants[194]. Ce traitement nécessite une réévaluation constante et doit être arrêté dès que possible. Par ailleurs, les effets indésirables de ces traitements sont nombreux[201] chez ces patients : ils majorent le risque d'accident vasculaire cérébral et entraînent une mortalité accrue. L'arrêt de ces traitements en dehors des phases d'agitation aiguë ne semble pas aggraver les troubles du comportement[202].
- Les hypnotiques et anxiolytiques peuvent aggraver les troubles cognitifs[203]. Ils doivent donc être utilisés avec précaution.
Traitements spécifiques des démences
Inhibiteurs de l'acétylcholinestérase
Ils inhibent la dégradation de l'acétylcholine, une molécule permettant la transmission entre certains neurones du cerveau par l'intermédiaire de ses synapses. Ainsi, ils visent à corriger le déficit en acétylcholine observé dans le cerveau des personnes atteintes de cette maladie.
Plusieurs inhibiteurs ont été testés de façon rigoureuse et ont prouvé une certaine efficacité, dans les formes légères à modérément sévères : le donépézil[204], la rivastigmine, et la galantamine. En 2007 la Commission française de la transparence a réévalué quatre anticholinestérasiques et a conclu à une ASMR mineure[205]. D'après la revue Prescrire leurs effets sont modestes, de quelques mois, chez environ 10 % des patients[206].
Bien que modestes, leurs effets sont significativement supérieurs à ceux du placebo : ralentissement ou retard du déclin cognitif et de la perte d'autonomie.
L'effet de ces traitements est stabilisateur et ils ne permettent pas de guérir la maladie, ni de récupérer le niveau de performances préexistant à sa survenue. Leurs utilisations exposent à de nombreuses interactions médicamenteuses, ainsi qu'à des effets indésirables.
Les anticholinestérasiques ont des effets secondaires[207], surtout de type digestif (nausées et vomissement). Certains induiraient une surmortalité cardiovasculaire et des tremblements et/ou une aggravation de symptômes parkinsoniens[208] ce qui a été à l'origine de controverses portant notamment sur leur justification économique[209]. Néanmoins, les organismes d'expertise les plus sérieux reconnaissent leur intérêt[210]. Les grandes agences de santé, en France, la Haute Autorité de Santé, ne recommandent plus leur utilisation dans la maladie d'Alzheimer en dehors d'un cadre très précis[211].
Antagonistes du NMDA
- Les récepteurs neuronaux au N-methyl-D-aspartate (NMDA) jouent un rôle important dans les processus de mémorisation. Il semble que lors de la maladie d'Alzheimer ces récepteurs soient hyperstimulés par le glutamate, ce qui serait délétère. Elle est réservée aux stades moyens ou avancés[212].
- La vitamine D a démontré une efficacité en synergie avec la mémantine. La mémantine et la vitamine D isolément n'ont guère amélioré l'état de patients atteints de la maladie d'Alzheimer mais ont permis des améliorations cognitives significatives en six mois lorsque prises conjointement[213].
Recherche
Modèles animaux
Afin d'étudier l'apparition de la maladie, des souris transgéniques sont utilisées pour reproduire les symptômes observés chez l'homme. Les mutations sont donc principalement effectuées sur les gènes de la protéine tau et/ou de la protéine amyloïde. Cependant, les résultats sur ces modèles animaux restent difficiles à interpréter, notamment pour l'efficacité des éventuels traitements testés et leur transfert vers l'homme[réf. nécessaire].
Les souris ayant eu une mutation sur le gène codant la protéine Tau montrent une apparition de la maladie peu prononcée et les souris ayant eu une mutation sur le gène codant la protéine amyloïde se comportent comme des souris saines. C'est seulement lorsque les deux gènes sont mutés que les souris développent une maladie semblable à celle d'Alzheimer[réf. nécessaire]. Cela ne se passe pas obligatoirement de façon identique chez l'homme, mais cela montrerait que les plaques amyloïdes potentialisent l'apparition de la maladie. Les neurofibrilles apparaissent dans un premier temps et lorsque les plaques amyloïdes apparaissent, la maladie se déclenche. Il est certain que chez l'homme, le processus pathologique se développe bien avant que les premiers signes cliniques n'apparaissent[réf. nécessaire].
Diagnostic
Le but des recherches diagnostic est de permettre un diagnostic plus sensible et plus spécifique de la maladie d'Alzheimer que l'interrogatoire et les tests neuropsychologiques. De meilleurs tests pourraient permettre peut-être un diagnostic plus précoce et un traitement plus précoce pour freiner l'évolution. L’intérêt de détecter précocement la maladie d'Alzheimer pourrait permettre aux personnes atteintes de bénéficier de traitements spécifiques plus précoces. De nouveaux critères, provenant d’une combinaison entre tests de mémoire, données d'imagerie cérébrale et marqueurs biologiques, pourraient permettre de détecter la maladie d'Alzheimer à un stade précoce, dès les premiers symptômes, « avec un taux de certitude diagnostique supérieur à 90 %. »[214] [réf. incomplète]
Projets de calcul distribué
Le projet international neuGRID est un système d'analyse des images d'imageries cérébrale. Il est financé par l'Union européenne, prévoit le développement d’une infrastructure numérique pour la recherche scientifique, fondée sur le système Grid. Il est équipé d’une interface d’utilisation facile, qui permettra aux chercheurs européens de neurosciences de faire avancer la recherche pour l’étude de la maladie d'Alzheimer et d'autres maladies neurodégénératives.
Traitements
Immunothérapie et vaccin
Aucun vaccin ou médicament immunogénique n'est aujourd'hui commercialisé. Un vaccin pouvant soigner cette maladie semble envisageable d'après des études menées chez l'animal. Les premiers tests sur l'homme ont été très décevants avec des effets secondaires majeurs (décès observés sans empêcher l'évolution de la maladie d'Alzheimer).
L'idée date de 1999 ; Dale Schenk, un chercheur américain, présentait dans la revue Nature une méthode semblant guérir la maladie chez des souris. En immunisant contre le peptide A bêta des souris transgéniques qui le surexpriment, il arrivait à prévenir l'apparition de dépôts chez les animaux jeunes et à limiter et même réduire leur extension chez les individus âgés.
Un premier essai clinique de phase 1 chez l'homme conduit ensuite en Angleterre a permis l'analyse suivante : les 80 patients traités supportent bien la vaccination et le quart d'entre eux produisent bien des anticorps. Un second essai a été interrompu en raison d'effets indésirables graves (méningoencéphalites). Le suivi ultérieur des patients qui ont reçu le vaccin est plus mitigé : même si chez certains patients traités les dépôts amyloïdes intracérébraux sont moins importants, le vaccin n'a pas empêché la progression de la détérioration intellectuelle jusqu'au stade terminal[215].
Un autre espoir, porté par le Japonais Tohru Hasegawa[216] est d'utiliser l'acide homocystéique comme cible d'un vaccin. Cet acide - chez des souris 3xTg-AD (transgéniques, modifiées de manière à mimer les symptômes de la maladie humaine) - semble en effet nécessaire à la progression de la dégénérescence typique de cette affection[217],[218].
Le taux de cet acide est plus élevé dans le cerveau des souris 3xTg-AD de 4 mois que chez les souris témoins normales. Quand des souris 3xTg-AD sont soumises à une carence en vitamine B6 (ce qui augmente la quantité d'acide homocystéique dans leur cerveau), cela aggrave aussi leurs troubles mémoriels, sauf en cas d'injection d'anticorps anti-acide homocystéique. Injecter ces mêmes anticorps à des souris 3xTG-AD plus vieilles et normalement alimentées a également un effet curatif, en tous cas pour les troubles mémoriels. Les chercheurs restent prudents, car il a été vu dans le passé que la souris n'était pas un modèle parfait pour cette maladie. Le vaccin devrait être testé sur des singes avant tout essai clinique sur l'homme.
Champs magnétiques
D'autres pistes sont en cours d'évaluation. Selon une étude[219], des souris (normales, et transgéniques présentant des troubles jugés comparables à ceux induits par la maladie d'Alzheimer) exposés plusieurs mois à certaines ondes électromagnétiques (de type téléphone portable ; 918 MHz ; 0,25 W/kg) améliorent leur mémoire, perdent moins de capacité cognitive en vieillissant et produisent moins de plaques amyloïdes dans leur hippocampe (-35 %) et leur cortex entorhinal (-32 %). Une température cérébrale plus élevée de 1 °C et une accélération du débit sanguin cérébral sont constatées, mais le mécanisme global n'est pas compris. Si un effet similaire était constaté chez l'Homme, une piste nouvelle de traitement, non médicamenteuse et non chirurgicale s'ouvrirait.
La voie du cholestérol cérébral
Un nouvel axe thérapeutique est envisagé par une équipe française de l'Inserm, dirigée par le Dr Nathalie Cartier, qui a montré que le cholestérol cérébral, quand il est en excès, était impliqué dans le développement de la maladie. Leur stratégie consiste donc à sur-exprimer l'enzyme CYP46A1 responsable de la dégradation du cholestérol cérébral, par thérapie génique. Les résultats sur les souris se sont avérés très prometteurs[220].
Épidémiologie
Deux formes de la maladie d'Alzheimer sont séparées :
- la forme familiale, plus précoce, d'origine principalement génétique et donc assez rare ;
- la forme sporadique, forme la plus répandue de la maladie et dont le risque augmente fortement avec l'âge.
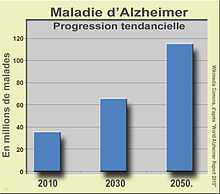
Dans le monde, le nombre de cas de malades d'Alzheimer est passé de 11 millions en 1980 à 18 millions en 2000 et 25 millions en 2004[222]. Il est estimé que 35,6 millions de personnes vivent désormais avec une maladie d'Alzheimer dans le monde. Elles seront 65,7 millions en 2030 et 115,4 millions en 2050[221]. À l'échelle mondiale, la maladie d’Alzheimer est la troisième cause d'invalidité pour les plus de 60 ans (après les atteintes de la moelle épinière et les cancers en phase terminale[13]) avec une prévalence de l'ordre de 4 à 6 % à cet âge[13]. Ces chiffres proviennent essentiellement des études épidémiologiques effectuées dans les pays développés, en effet bien que cette maladie s'observe sur tous les continents, elle est peu caractérisée dans les pays en voie de développement où l'espérance de vie est souvent plus courte et les enquêtes épidémiologiques plus rares. La maladie d'Alzheimer est cependant considérée comme une pandémie[192].
À part l'âge, les facteurs de risque génétique évoqués plus haut comme l'ApoE-e4, la petite taille, le tabagisme, certaines pathologies pré-existantes (diabète, hypertension, taux de cholestérol élevé)[223], l'alimentation (viande et sucre) et l'inactivité physique, n'expliquent pas entièrement les variations de prévalence constatées, et les autres facteurs de risque de la maladie sont mal connus. Les chercheurs se tournent de plus en plus vers la recherche de causes environnementales[224].
Dans ce but ils examinent les prévalences de la maladie par pays[225],[226] - les pays nordiques européens et les Etats-Unis sont les plus touchés -, ou par état aux Etats-Unis[227] où l'état de Washington est inexplicablement le plus touché.
L’incidence — aux mêmes âges — est toujours plus forte dans les pays riches qu'ailleurs (sauf au Japon où l'incidence est très faible, et moindrement en Amérique centrale et du Sud). Ceci est vrai pour les populations urbaines supposées plus exposées aux pollutions, mais aussi pour les populations rurales (qui par exemple en Inde développent 5,4 fois moins d'Alzheimer qu'en Pennsylvanie[228]).
De même, les Afro-Américains vivant aux États-Unis sont beaucoup plus touchés que les Yorubas du Nigeria. Des chercheurs ont comparé l'incidence de Maladie d'Alzheimer (MA) dans une population Yoruba du Nigeria et chez des Afro-Américains génétiquement proches (fréquence comparable (26 à 29 %) et élevée d'allèles APOE4)[229]. Le risque de maladie d’Alzheimer pour ces individus est deux fois moindre au Nigeria (1,15 %) qu'en Amérique du Nord (2,52 %) à âge égal, ce qui plaide aussi pour une cause environnementale, au moins dans 50 % des cas[230].
Cette maladie est plus rare en Asie[231]. Le Japon est notamment une exception parmi les pays industrialisés et riches. La prévalence de la maladie y est presque 10 fois plus faible qu'en France. Cependant, un Japonais vivant à Hawaï ou aux États-Unis voit son niveau de risque augmenter (5,4 % pour les Japonais d’Hawaii) et se rapprocher de celui d'un Américain moyen, d'un Caucasien ou Européen[231],[232]). De même, 5,7 % des Japonais ayant émigré au Brésil ont après quelques décennies le même risque de développer la maladie qu'un Brésilien moyen[233],[234]. Cette analyse montre, comme pour l'étude sur les populations nigérianes, la prépondérance de l'environnement dans le déclenchement de la maladie d’Alzheimer. Au Japon, l'Alzheimer est rare, mais la démence vasculaire est - comme aux États-Unis - très élevée, probablement en raison d'une consommation trop importante de sel. Cette maladie diminue au Japon grâce à la prévention et à une prise en charge plus efficace de l'hypertension[235].
Aux États-Unis, pays très touché, 5,3 millions de personnes ont la maladie d'Alzheimer[236], avec une prévalence double chez les africains-américains et chez les hispaniques, par rapport à la population d'origine anglo-saxonne.
Impact économique
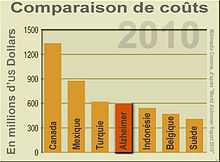
La maladie d'Alzheimer était, en 2010, dans les pays développés, l'une des maladies les plus coûteuses : 604 milliards de dollars. Aux États-Unis, elle a couté 94 milliards de dollars à l'assurance maladie américaine en 2008[237].
Perspectives en Europe
En Europe, l’incidence des démences devrait croître en 50 ans de 1,9 million de nouveaux cas par an à 4,1 millions, selon les scenarii[238].
En Belgique, 5 à 10 % des plus de 65 ans sont touchés et près de 20 % des plus de 80 ans[239].
En France, l'étude « PAQUID » (1988-2001) a fait ressortir que 17,8 % des personnes de plus de 75 ans sont atteintes de la maladie d'Alzheimer ou d'un syndrome apparenté. D'après une évaluation ministérielle de 2004, environ 860 000 personnes seraient touchées par la maladie d’Alzheimer en France. Un chiffre qui pourrait atteindre 1,3 million en 2020 et 2,1 millions en 2040. Le nombre de nouveaux cas est d'environ 225 000 par an[240]. La prévalence des démences chez les plus de 75 ans atteint presque 18 % (maladie d’Alzheimer à 80 %)[241]. Ceci représente environ 900 000 malades (et dans les années 2000 environ 220 000 nouveaux cas par an), avec des tendances et projections très alarmantes : 1 200 000 malades en 2020, et plus de 2 000 000 vers 2040[242][réf. incomplète].
Histoire et société
Histoire

Dès l'antiquité, philosophes et médecins associèrent l'âge avancé à une augmentation de la démence[1].
Ce n'est qu'en 1901, qu'Alois Alzheimer (1864-1915), un psychiatre et neuropathologiste allemand, identifia le premier cas de la maladie qui portera son nom, chez une patiente de 50 ans, Auguste Deter. Il suivit son cas jusqu'à sa mort en 1906 et décrivit les altérations anatomiques observées sur son cerveau. Durant les cinq années suivantes, onze cas similaires furent rapportés dans la littérature médicale, certains employant déjà le terme de maladie d'Alzheimer[243].
Les caractéristiques particulières de la maladie furent isolées pour la première fois par le psychiatre Emil Kraepelin (1856-1926), après le retrait de certains symptômes présents dans le cas initial d'Auguste D. (délire, hallucination et artériosclérose)[244]. Il inclut cette toute nouvelle maladie d'Alzheimer, qu'il appela également démence pré-sénile, comme un sous-type des démences séniles dans son manuel de psychiatrie publié en 1910[245].
Durant la majeure partie du XXe siècle, le diagnostic de maladie d'Alzheimer fut réservé aux individus âgés de 45 à 65 ans qui développaient des démences. La terminologie changea en 1977, lors d'une conférence sur la maladie d’Alzheimer, où il fut conclu que les manifestations cliniques et pathologiques des démences séniles et pré-sénile étaient identiques, bien que les auteurs n’exclurent pas qu'elles aient des origines différentes[246]. Cela aboutit à un diagnostic de Maladie d'Alzheimer indépendant de l'âge[247]. Le terme de démence sénile de type Alzheimer fut utilisé durant un certain temps pour décrire les cas d'Alzheimer supérieur à 65 ans, et l’Alzheimer classique ceux plus jeunes. Finalement, le terme unique de maladie d'Alzheimer fut formellement adopté dans la nomenclature médicale pour décrire les individus de tout âge présentant un ensemble particulier de symptômes, de progression dans le temps et de caractéristiques neuropathologiques[248].
L'après-baby boom (ou papy boom), la maitrise de la fécondité et le progrès médical conduisent les sociétés à devoir vivre une période où les personnes âgées seront très nombreuses. Cette maladie fait donc l'objet d'une attention particulière, notamment en France avec l'observatoire national sur la recherche sur la maladie d'Alzheimer (ONRA)[249].
En 2009, la maladie d'Alzheimer correspond à plus de la moitié des cas de démence de la personne âgée dans les pays riches.
Personnes célèbres décédées de la maladie d'Alzheimer
Voir la catégorie « Mort de la maladie d'Alzheimer »
Médias
Œuvres cinématographiques
- 2001 : Iris de Richard Eyre
- 2001 : Se souvenir des belles choses de Zabou Breitman
- 2002 : La Maladie de la mémoire de Richard Dindo
- 2004 : La Mémoire du tueur (De zaak Alzheimer) de Erik Van Looy
- 2004 : N'oublie jamais (The Notebook) de Nick Cassavetes
- 2004 : A Moment to Remember de John H. Lee
- 2005 : Une grande bouffée d'amour ! (Aiguemarine C°)
- 2005 : Black de Sanjay Leela Bhansali
- 2006 : Memories of Tomorrow de Yukihiko Tsutsumi
- 2007 : Loin d'elle (Away from Her) de Sarah Polley
- 2007 : La Brunante de Fernand Dansereau
- 2007 : Ta mémoire… Mon Amour ! (Aiguemarine C°)
- 2008 : Cortex de Nicolas Boukhrief
- 2008 : U, Me Aur Hum de Ajay Devgan
- 2009 : Je me souviens mieux quand je peins, documentaire d'Eric Ellena et Berna Huebner
- 2009 : La Verticalidad del Tiempo, documentaire de Raùl Fernandez Rincon et Marianne Ruiz, Luca Films, Madrid
- 2010 : The Secret Life of the American Teenager, Saison 1, épisode 8/23 (La Visite), série TV de Keith Truesdell
- 2010 : L'Absence de Cyril de Gasperis
- 2010 : Le Monde de Barney de Richard J. Lewis
- 2010 : Poetry de Lee Chang-Dong
- 2011 : Je n'ai rien oublié, de Bruno Chiche, d'après le roman Small World de Martin Suter
- 2011 : La Planète des singes : Les Origines de Rupert Wyatt
- 2011 : A Thousand Days' Promise ou Forget me not, œuvre coréenne de Jung Eul-young
- 2014 : Still Alice de Wash Westmoreland (en)
Œuvres littéraires
Depuis 1997, on note une forte augmentation des œuvres comportant des personnages atteints de la maladie d'Alzheimer ou de maladie apparentée[250].
- Still Alice, de Lisa Genova, Pocket Books 2007
- Alzheimer, mode d'emploi, de Jean-Pierre Polydor, L'esprit du temps 2009
- Histoire de ma mère, de Yasushi Inoué, Stock 1997, 2004 et 2007
- Je ne suis pas sortie de ma nuit, de Annie Ernaux, Gallimard 1997 et Folio 1999
- Ton chapeau au vestiaire, de Nadine Trintignant, Fayard 1997 et Pocket 1999
- Small World, de Martin Suter, Christian Bourgois 1998, et Points Seuil 2000
- Quel jour sommes-nous ? de Firmin Le Bourhis, Chiron 2000
- La Cavale du géomètre, d'Arto Paasilinna, Gallimard/folio 2000
- Élégies pour Iris, de John Bailey (en), L’Olivier 2001
- Les jours heureux, de Laurent Graff, La Dilettante 2001 et J’ai Lu 2003
- L’Effacement de l’aube, de Ronald Nossintchouk, E-Dite 2002
- Les Corrections, de Jonathan Franzen, L’Olivier 2002 et Points Seuil 2003
- Pourquoi ma mère me rend folle, de Françoise Laborde, Ramsay 2002 et J’ai Lu 2003
- Ma mère n’est pas un philodendron, de Françoise Laborde, Fayard 2003 et J’ai Lu 2005
- L’Éclipse, de Serge Rezvani, Actes Sud 2003
- Ultime amour, de Serge Rezvani, Les Belles Lettres 2012
- Mon vieux, de Thierry Jonquet, Seuil 2004
- Le Dernier qui part ferme la maison, de Michèle Fitoussi, Grasset 2004 et Poche 2006
- Les Cœurs décousus, de Jacqueline Girard-Frésard, Le Cherche Midi 2004
- Puzzle : journal d’une Alzheimer, de Claude Couturier, Josette Lyon 2004
- Les Quantités Négligeables (Le combat ordinaire, Tome 2), de Manu Larcenet, Dargaud 2004
- Sœurs, de Catherine Locandro, Gallimard 2005
- Mémorial, de Cécile Wajsbrot, Zulma 2005
- Le Piano désaccordé, de Christine Devars, Anne Carrière 2005
- Devant ma mère, de Pierre Pachet, Gallimard 2007
- Rides, de Paco Roca, Delcourt 2007
- Élégie : 1996-2006. La maladie d’Alzheimer vécue à deux, de Jean Sauvy, L’harmattan 2007
- Les Artistes de la mémoire, de Jeffrey Moore, Philippe Rey, 2007
- Les Madones de Léningrad, de Debra Dean, Grasset, 2007
- Le vieux roi en son exil : récit, de Arno Geiger, Gallimard (Du monde entier), 2012
- La mémoire de ma mère Annie Girardot, de Giulia Salvatori, M. Lafon 2007
- Rainbows end, de Vernor Vinge, Robert Laffont 2007
- On n’est pas là pour disparaître, de Olivia Rosenthal, Gallimard (Verticales) 2007
- La Vie sur terre, de Dorothée Janin, Denoël 2007
- La Plume du silence - Toi et moi… et Alzheimer, de Jean Witt, Presses de la Renaissance 2007
- L'Homme inquiet de Henning Mankell, Seuil Policier 2010
- De Peur que j'oublie, de Marie-Noël Rio, Les Éditions du Sonneur, 2014
Notes et références
- 1 2 (en) NC Berchtold et CW Cotman, « Evolution in the conceptualization of dementia and Alzheimer's disease: Greco-Roman period to the 1960s », Neurobiol. Aging, vol. 19, no 3, , p. 173–189. (PMID 9661992, DOI 10.1016/S0197-4580(98)00052-9)
- ↑ (en) « About Alzheimer's Disease: Symptoms », National Institute on Aging (consulté le 28 décembre 2011)
- ↑ (en) R. Brookmeyer, S. Gray et C. Kawas, « Projections of Alzheimer's disease in the United States and the public health impact of delaying disease onset », American Journal of Public Health, vol. 88, no 9, , p. 1337–1342. (PMID 9736873, PMCID 1509089, DOI 10.2105/AJPH.88.9.1337)
- ↑ Alzheimer, espérance de vie et âge au moment du diagnostic, sur le site caducee.net du 21 novembre 2002.
- ↑ (en) Mölsä PK, Marttila RJ, Rinne UK, « Survival and cause of death in Alzheimer's disease and multi-infarct dementia », Acta Neurol Scand, vol. 74, no 2, , p. 103–107. (PMID 3776457, DOI 10.1111/j.1600-0404.1986.tb04634.x)
- 1 2 (en) G Waldemar, B Dubois, M Emre, J. Georges, I. G. McKeith, M. Rossor, P. Scheltens, Tariska et B. Winblad, « Recommendations for the diagnosis and management of Alzheimer's disease and other disorders associated with dementia: EFNS guideline », Eur J Neurol, vol. 14, no 1, , e1–26. (PMID 17222085, DOI 10.1111/j.1468-1331.2006.01605.x)
- ↑ (en) Tabert MH, Liu X, Doty RL, Serby M, Zamora D, Pelton GH, Marder K, Albers MW, Stern Y, Devanand DP, « A 10-item smell identification scale related to risk for Alzheimer's disease », Ann Neurol, vol. 58, no 1, , p. 155–160. (PMID 15984022, DOI 10.1002/ana.20533)
- ↑ [PDF] (en) « More research needed on ways to prevent Alzheimer's, panel finds », National Institute on Aging, (consulté le 29 février 2008)
- ↑ [PDF] (en) « The MetLife study of Alzheimer's disease: The caregiving experience » [PDF], MetLife Mature Market Institute, (consulté le 5 février 2011)
- ↑ (en) Thompson CA, Spilsbury K, Hall J, Birks Y, Barnes C, Adamson J, « Systematic review of information and support interventions for caregivers of people with dementia », BMC Geriatr, vol. 7, , p. 18 (PMID 17662119, PMCID PMC1951962, DOI 10.1186/1471-2318-7-18)
- ↑ (en) Schneider J, Murray J, Banerjee S, Mann A, « EUROCARE: a cross-national study of co-resident spouse carers for people with Alzheimer's disease: I—Factors associated with carer burden », International Journal of Geriatric Psychiatry, vol. 14, no 8, , p. 651–661. (PMID 10489656).
- ↑ (en) Murray J, Schneider J, Banerjee S, Mann A, « EUROCARE: a cross-national study of co-resident spouse carers for people with Alzheimer's disease: II—A qualitative analysis of the experience of caregiving », International Journal of Geriatric Psychiatry, vol. 14, no 8, , p. 662–667. (PMID 10489657)
- 1 2 3 (en) Cleusa P. Ferri, Martin Prince, Carol Brayne, Henry Brodaty, Laura Fratiglioni, Mary Ganguli, Kathleen Hall, Kazuo Hasegawa, Hugh Hendrie, Yueqin Huang, Anthony Jorm, Colin Mathers, Paulo R Menezes, Elizabeth Rimmer, Marcia Scazufca, « Global prevalence of dementia: a Delphi consensus study », Alzheimer's Disease International, The Lancet, vol. 366, no 9503, 17 décembre 2005-6 janvier 2006, p. 2112-2117. (DOI 10.1016/S0140-6736(05)67889-0, lire en ligne [PDF])
- ↑ (en) R Brookmeyer, E Johnson, K Ziegler-Graham et HM Arrighi, « Forecasting the global burden of Alzheimer's disease », Alzheimer's and Dementia, vol. 3, no 3, , p. 186–191. (PMID 19595937, DOI 10.1016/j.jalz.2007.04.381, lire en ligne)
- ↑ Bonin-Guillaume S, Zekry D, Giacobini E, Gold G, Michel JP, « Impact économique de la démence », Presse Med, vol. 34, no 1, , p. 35–41. (ISSN 0755-4982, PMID 15685097)
- ↑ (en) PD Meek, K McKeithan et GT Schumock, « Economic considerations in Alzheimer's disease », Pharmacotherapy, vol. 18, no 2 Pt 2, , p. 68–73; discussion 79–82. (PMID 9543467)
- 1 2 3 (en) Bäckman L, Jones S, Berger AK, Laukka EJ, Small BJ, « Multiple cognitive deficits during the transition to Alzheimer's disease », J Intern Med, vol. 256, no 3, , p. 195–204. (PMID 15324363, DOI 10.1111/j.1365-2796.2004.01386.x)
- ↑ (en) Nygård L, « Instrumental activities of daily living: a stepping-stone towards Alzheimer's disease diagnosis in subjects with mild cognitive impairment? », Acta Neurol Scand, vol. Suppl, no 179, , p. 42–46. (PMID 12603250, DOI 10.1034/j.1600-0404.107.s179.8.x)
- 1 2 (en) Arnáiz E, Almkvist O, « Neuropsychological features of mild cognitive impairment and preclinical Alzheimer's disease », Acta Neurol Scand, vol. 107, no Suppl. s179, , p. 34–41. (PMID 12603249, DOI 10.1034/j.1600-0404.107.s179.7.x)
- ↑ (en) Landes AM, Sperry SD, Strauss ME, Geldmacher DS, « Apathy in Alzheimer's disease », J Am Geriatr Soc, vol. 49, no 12, , p. 1700–1707. (PMID 11844006, DOI 10.1046/j.1532-5415.2001.49282.x)
- ↑ (en) Petersen RC, « The current status of mild cognitive impairment—what do we tell our patients? », Nat Clin Pract Neurol, vol. 3, no 2, , p. 60–61. (PMID 17279076, DOI 10.1038/ncpneuro0402)
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 (en) Förstl H, Kurz A, « Clinical features of Alzheimer's disease », European Archives of Psychiatry and Clinical Neuroscience, vol. 249, no 6, , p. 288–290. (PMID 10653284, DOI 10.1007/s004060050101)
- ↑ A.-M. Ergis, E. Eusop-Roussel, « Les troubles précoces de la mémoire épisodique dans la maladie d’Alzheimer [Early episodic memory impairments in Alzheimer's disease] » Revue Neurologique, mai 2008;164(Supplément 3):S96–S101. PMID 18675054
- ↑ (en) Carlesimo GA, Oscar-Berman M, « Memory deficits in Alzheimer's patients: a comprehensive review », Neuropsychol Rev, vol. 3, no 2, , p. 119–169. (PMID 1300219, DOI 10.1007/BF01108841)
- ↑ (en) Jelicic M, Bonebakker AE, Bonke B, « Implicit memory performance of patients with Alzheimer's disease: a brief review », International Psychogeriatrics, vol. 7, no 3, , p. 385–392. (PMID 8821346, DOI 10.1017/S1041610295002134)
- 1 2 (en) Taler V, Phillips NA, « Language performance in Alzheimer's disease and mild cognitive impairment: a comparative review », J Clin Exp Neuropsychol, vol. 30, no 5, , p. 501–556. (PMID 18569251, DOI 10.1080/13803390701550128)
- 1 2 3 (en) Frank EM, « Effect of Alzheimer's disease on communication function », J S C Med Assoc, vol. 90, no 9, , p. 417–423. (PMID 7967534)
- ↑ (en) Volicer L, Harper DG, Manning BC, Goldstein R, Satlin A, « Sundowning and circadian rhythms in Alzheimer's disease », Am J Psychiatry, vol. 158, no 5, , p. 704–711. (PMID 11329390, DOI 10.1176/appi.ajp.158.5.704, lire en ligne)
- ↑ (en) Gold DP, Reis MF, Markiewicz D, Andres D, « When home caregiving ends: a longitudinal study of outcomes for caregivers of relatives with dementia », J Am Geriatr Soc, vol. 43, no 1, , p. 10–16. (PMID 7806732)
- ↑ (en) Wenk GL, « Neuropathologic changes in Alzheimer's disease », J Clin Psychiatry, vol. 64 Suppl 9, , p. 7–10. (PMID 12934968)
- ↑ (en) Desikan RS, Cabral HJ, Hess CP, « Automated MRI measures identify individuals with mild cognitive impairment and Alzheimer's disease », Brain, vol. 132, no Pt 8, , p. 2048–2057. (PMID 19460794, PMCID 2714061, DOI 10.1093/brain/awp123)
- ↑ (en) Moan R, « MRI software accurately IDs preclinical Alzheimer's disease », Diagnostic Imaging, (lire en ligne)
- ↑ (en) Is Alzheimer’s disease akin to type 3 diabetes?, sur le site bostonglobe.com du 8 octobre 2012.
- ↑ (en) de la Monte SM, Wands JR, « Alzheimer's disease is type 3 diabetes-evidence reviewed », J Diabetes Sci Technol, vol. 2, no 6, , p. 1101-13. (PMID 19885299, PMCID PMC2769828, lire en ligne [PDF])
- ↑ (en) Delacourte A, David JP, Sergeant N et al. « The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer's disease » Neurology 1999;52:1158-65.
- ↑ (en) Delacourte A, Buée L. « Normal and pathological Tau proteins as factors for microtubule assembly » Int Rev Cytol. 1997;171:167-224.
- 1 2 3 (en) Blennow K, de Leon MJ, Zetterberg H, « Alzheimer's disease », Lancet, vol. 368, no 9533, , p. 387–403. (PMID 16876668, DOI 10.1016/S0140-6736(06)69113-7)
- 1 2 3 (en) Waring SC, Rosenberg RN, « Genome-wide association studies in Alzheimer disease », Arch Neurol, vol. 65, no 3, , p. 329-334. (PMID 18332245, DOI 10.1001/archneur.65.3.329)
- 1 2 (en) Selkoe DJ, « Translating cell biology into therapeutic advances in Alzheimer's disease », Nature, vol. 399, no 6738 Suppl, , A23–31. (PMID 10392577, DOI 10.1038/19866)
- ↑ (en) Shioi J, Georgakopoulos A, Mehta P, Zen Kouchi, Claudia M. Litterst, Lia Baki et Nikolaos K. Robakis, « FAD mutants unable to increase neurotoxic Aβ 42 suggest that mutation effects on neurodegeneration may be independent of effects on Abeta », J Neurochem, vol. 101, no 3, , p. 674–681. (PMID 17254019, DOI 10.1111/j.1471-4159.2006.04391.x)
- ↑ (en) Strittmatter WJ, Saunders AM, Schmechel D, M Pericak-Vance, J Enghild, GS Salvesen et AD Roses, « Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease », Proc. Natl. Acad. Sci. USA, vol. 90, no 5, , p. 1977–1981. (PMID 8446617, PMCID 46003, DOI 10.1073/pnas.90.5.1977)
- 1 2 (en) Mahley RW, Weisgraber KH, Huang Y, « Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer's disease », Proc. Natl. Acad. Sci. U.S.A., vol. 103, no 15, , p. 5644–5651. (PMID 16567625, PMCID 1414631, DOI 10.1073/pnas.0600549103)
- ↑ (en) Hall K, Murrell J, Ogunniyi A, Deeg M, Baiyewu O, Gao S, Gureje O, Dickens J, Evans R, Smith-Gamble V, Unverzagt FW, Shen J, Hendrie H, « Cholesterol, APOE genotype, and Alzheimer disease: An epidemiologic study of Nigerian Yoruba », Neurology, vol. 66, no 2, , p. 223–227. (PMID 16434658, PMCID 2860622, DOI 10.1212/01.wnl.0000194507.39504.17, lire en ligne)
- ↑ (en) Gureje O, Ogunniyi A, Baiyewu O, Price B, Unverzagt FW, Evans RM, Smith-Gamble V, Lane KA, Gao S, Hall KS, Hendrie HC, Murrell JR, « APOE ε4 Is Not Associated with Alzheimer's Disease in Elderly Nigerians », Ann Neurol, vol. 59, no 1, , p. 182–185. (PMID 16278853, PMCID 2855121, DOI 10.1002/ana.20694, lire en ligne)
- ↑ (en) Jonsson T, Stefansson H, Steinberg S et al. « Variant of TREM2 associated with the risk of Alzheimer's disease » N Engl J Med. 2013;368:107-116.
- ↑ (en) Guerreiro R, Wojtas A, Bras J et al. « TREM2 variants in Alzheimer's disease » N Engl J Med. 2013;368:117-127.
- ↑ (en) Takahashi K, Rochford CD, Neumann H, « Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2 » J Exp Med. 2005;201:647-657.
- ↑ (en) Neumann H, Daly MJ. « Variant TREM2 as risk factor for Alzheimer's disease » N Engl J Med. 2013;368:182-184.
- ↑ LA TRISOMIE 21, sur le site alzheimer-adna.com du 21 mars 2006.
- ↑ Trisomie 21 (31c), sur le site ujf-grenoble.fr de novembre 2005.
- ↑ Maladie d'Alzheimer : Épidémiologie, génétique et hypothèses physiopathologiques, sur le site inist.fr.
- ↑ (en) J.-C. Lambert et al. « Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease » Nature Genetics vol. 41, p. 1094-1099, 2009 (Résumé).
- ↑ (en) D. Harold et al. « Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease » Nature Genetics vol. 41, p. 1088-1093, 2009 (Résumé).
- ↑ Grégoria Kalpouzos, Francis Eustache, Béatrice Desgranges Réserve cognitive et fonctionnement cérébral au cours du vieillissement normal et de la maladie d’Alzheimer, Inserm, Psychologie et NeuroPsychiatrie du vieillissement. Volume 6, Numéro 2, p. 97-105, juin 2008, Article intégral.
- ↑ Recommandations de la HAS[Qui ?], page 2.
- ↑ Étude épidémiologique française ayant porté sur 3 000 sujets suivis durant 14 ans, H. Amieva et al. Ann Neurol. 2008 64;492.
- 1 2 3 Recommandations HAS 2011[PDF], sur le site has-sante.fr.
- ↑ Un test ultra-rapide pour évaluer les fonctions cognitives des sujets âgés, sur le site testcodex.org.
- ↑ Un test ultra-rapide pour évaluer les fonctions cognitives des sujets âgés, sur le site testcodex.org.
- ↑ Test de l'horloge, sur le site ammppu.org.
- ↑ Le test de 5 mots, sur le site esculape.com.
- ↑ Test, sur le site mocatest.org.
- ↑ Test de Gröber et Buschke[PDF], sur le site cofemer.fr.
- ↑ (en) Ralph M. Reitan, « Validity of the Trail Making Test as an indicator of organic brain damage », Perceptual and Motor Skills, vol. 8, no 3, , p. 271-276. (ISSN 0031-5125, 1558-688X, DOI 10.2466/pms.1958.8.3.271, lire en ligne)
- ↑ Test de Stroop sur le site Psycho.univ-paris5.fr.
- ↑ Serge Bakchine, Marie-Odile Habert Classification des démences : aspects nosologiques, Médecine Nucléaire, Volume 31, Issue 6, juin 2007, pages 278–293.
- ↑ Height and Alzheimer's disease: findings from a case-control study
- ↑ Relationship between body height and dementia
- ↑ (en)Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K, et al. Midlife vascular risk factors and Alzheimer’s disease in later life: longitudinal, population based study. BMJ 2001;322:1447-51 (Résumé).
- ↑ (en)Whitmer RA, Sidney S, Selby J, Johnston SC, Yaffe K. Midlife cardiovascular risk factors and risk of dementia in late life. Neurology 2005;64:277-81 (Résumé).
- ↑ (en)Luchsinger JA, Reitz C, Honig LS, Tang MX, Shea S, Mayeux R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology 2005;65:545-51 (Résumé).
- ↑ (en)Newman AB, Fitzpatrick AL, Lopez O, Jackson S, Lyketsos C, Jagust W, et al. Dementia and Alzheimer’s disease incidence in relationship to cardiovascular disease in the Cardiovascular Health Study cohort. J Am Geriatr Soc 2005;53:1101-7.
- ↑ (en)Nien-Chen Li, Austin Lee, Rachel A Whitmer, Miia Kivipelto, Elizabeth Lawler, Lewis E Kazis & Benjamin Wolozin ; Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis ; BMJ 2010;340:b5465, doi:10.1136/bmj.b5465 (2010/01/12) ; résumé, publié en Creative Commons Attribution Non-commercial License, consulté 5 avril 2010).
- ↑ (en)De la Torre JC. Is Alzheimer’s disease a neurodegenerative or a vascular disorder ? Data, dogma, and dialectics. Lancet Neurol 2004;3:184-90.
- ↑ (en) Ruitenberg A, den Heijer T, Bakker SL, van Swieten JC, Koudstaal PJ, Hofman A, et al. « Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study » Ann Neurol. 2005;57:789-94.
- ↑ (en) Encinas M, De Juan R, Marcos A, Gil P, Barabash A, Fernandez C, et al. « Regional cerebral blood flow assessed with 99mTc-ECD SPET as a marker of progression of mild cognitive impairment to Alzheimer’s disease » Eur J Nucl Med Mol Imaging 2003;30:1473-80.
- ↑ (en) « Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis » BMJ 2010;340:b5465. PMID 20068258 DOI:10.1136/bmj.b5465
- ↑ (en) Sjogren M, Mielke M, Gustafson D, Zandi P, Skoog I. « Cholesterol and Alzheimer’s disease—is there a relation ? » Mech Ageing Dev. 2006;127:138-47.
- ↑ (en) Morris MC, Evans DA, Bienias JL, Tangney CC, Bennett DA, Aggarwal N, Schneider J, Wilson RS, « Dietary fats and the risk of incident Alzheimer disease », Arch Neurol, vol. 60, no 2, , p. 194-200. (PMID 12580703)
- ↑ (en) Rusanen M, Kivipelto M, Quesenberry Jr CP, Zhou J, Whitmer RA, « Heavy smoking in midlife and long-term risk of Alzheimer disease and vascular dementia » Arch Intern Med. 2011;171:333-339.
- ↑ L'abus de tranquillisants et de somnifères augmenterait le risque d'Alzheimer.
- ↑ Lien possible entre les benzodiazépines et la Maladie d'Alzheimer.
- ↑ Les anxiolytiques et somnifères augmenteraient le risque d'Alzheimer.
- ↑ (en)Glucose tolerance status and risk of dementia in the community, neurology.org.
- ↑ Glucose Levels and Risk of Dementia.
- ↑ Épidémiologie de la maladie d’Alzheimer Jean-François Dartigues, Claudine Berr, Catherine Helmer, Luc Letenneur Med Sci (Paris) 18 (6-7) 737-743 (2002).
- ↑ (en) Jorm AF. « Is depression a risk factor for dementia or cognitive decline : a review » Gerontology 2000; 46:219-227.
- ↑ (en) Fratiglioni L, Launer LJ, Andersen K, et al. « Incidence of dementia and major subtypes in Europe: a collaborative study of population-based cohorts » Neurology 2000;54(suppl 5):S10-5.
- ↑ Meta-analysis of modifiable risk factors for Alzheimer's disease
- ↑ (en) Mayeux R, Ottman R, Tang M, et al. « Genetic susceptibility and head injury as risk factors for Alzheimer’s disease among community-dwelling elderly persons and their first-degree relatives » Ann Neurol. 1993;33:494-501.
- ↑ (en) Li G, Shen Y, Li Y, Chen C, Zhau Y, Silverman J. « A case-control study of Alzheimer’s disease in China » Neurology 1992;42:1481-1488.
- ↑ (en) Martyn CN, Barker JP, Osmond C, Harris EC, Edwarson JA, Lacey RF. « Geographical relation between Alzheimer’s disease and aluminium in drinking water » Lancet 1989;14:59-62.
- ↑ (en) Martyn C, Coggon D, Lacey R. « Aluminium concentrations in drinking water and risk of Alzheimer’s disease » Epidemiology 1997;8:281-286.
- ↑ (en) Rondeau V, Commenges D, Jacqmin-Gadda H, Dartigues JF. « Relation between aluminum concentrations in drinking water and Alzheimer’s disease: An 8-year followup study » Am J Epidemiol. 2000;152:59-66.
- ↑ (en) Méta-analyse sur la relation entre l'inactivité physique et la maladie d'Alzheimer[PDF].
- ↑ « Facteurs environnementaux impliqués dans la maladie d’Alzheimer. Le mercure dentaire, probable déterminant majeur », lire en ligne sur em-consulte.com.
- ↑ (en) Health effects of dental amalgam exposure: a retrospective cohort study., M Bates et al. Int. J. Epidemiol. (2004) 33 (4): p. 894-902.
- ↑ (en) Does Inorganic Mercury Play a Role in Alzheimer’s Disease? A Systematic Review, J Mutter, J Alzheimers Dis. 2010; 22(2):357-74.
- ↑ OMS (WHO/IPCS) ; 1991 : extrait de Global Mercury Assessment (ONU; Mercury Programm, chap. 4). (lire en ligne).
- ↑ (en)Weiner JA, Nylander M. ; The relationship between mercury concentration in human organs and different predictor variables. Sci Total Environ. Septembre 1993 ; 0;138(1-3):101-15.
- ↑ (en)Nylander M, Friberg L, Lind B ; Mercury concentrations in the human brain and kidneys in relation to exposure from dental amalgam fillings. Swed Dent J. ; 1987 ; 11(5):179-87.
- ↑ (en)Eggleston DW, Nylander M J Prosthet ; Correlation of dental amalgam with mercury in brain tissue. Dent. ; décembre 1987 ; 58(6):704-7.
- ↑ (en)Drasch G, Schupp I, Hofl H, Reinke R, Roider G Eur ; Mercury burden of human fetal and infant tissues. ; J Pediatr. ; août 1994 ; 153(8):607-10.
- ↑ (en)Guzzi G, Grandi M, Cattaneo C, Calza S, Minoia C, Ronchi A, Gatti A, Severi G. Dental amalgam and mercury levels in autopsy tissues: food for thought ; Am J Forensic Med Pathol. 2006 Mar;27(1):42-5.
- ↑ (en)Mutter J, Naumann J, Sadaghiani C, Schneider R, Walach H. Alzheimer disease: mercury as pathogenetic factor and apolipoprotein E as a moderator ; Neuro Endocrinol Lett. octobre 2004 ; 25(5):331-9.
- ↑ (en)Pendergrass JC, Slevin JT, Haley BE. Duhr EF, HgEDTA complex inhibits GTP interactions with the E-site of brain beta-tubulin. ; Toxicol Appl Pharmacol. 1993 Oct;122(2):273-80.
- ↑ (en)Jones JA, Lavallee N, Alman J, Sinclair C, Garcia RI. ; Caries incidence in patients with dementia. Geriatric Dental Program, Department of Veterans Affairs Medical Center, Bedford, MA 01730, États-Unis. Gerodontology. 1993 Dec;10(2):76-82 (résumé).
- ↑ Rejnefelt I, Andersson P, Renvert S. (Département des sciences de la santé de l'Université de Kristianstad) Oral health status in individuals with dementia living in special facilities. Int J Dent Hyg. mai 2006 ; 4(2):67-71.
- ↑ (en)Pendergrass JC, Haley BE, Vimy MJ, Winfield SA, Lorscheider FL. Mercury vapor inhalation inhibits binding of GTP to tubulin in rat brain: similarity to a molecular lesion in Alzheimer diseased brain. ; Neurotoxicology. ; 1997 ;18(2):315-24.
- ↑ (en)Thompson CM, Markesbery WR, Ehmann WD, Mao YX, Vance DE Regional brain trace-element studies in Alzheimer's disease. Neurotoxicology. 1988 Spring;9(1):1-7.
- ↑ (en)Ehmann WD, Markesbery WR, Alauddin M, Hossain TI, Brubaker EH ; Brain trace elements in Alzheimer's disease ; Neurotoxicology. 1986 Spring;7(1):195-206.
- ↑ (en)Increased blood mercury levels in patients with Alzheimer's disease. Hock C, Drasch G, Golombowski S, Müller-Spahn F, Willershausen-Zönnchen B, Schwarz P, Hock U, Growdon JH, Nitsch RM. J Neural Transm. 1998;105(1):59-68.
- ↑ (en)Krauhs E, Little M, Kempf T, Hofer-Warbinek R, Ade W, Ponstingl H. ; Complete amino acid sequence of beta-tubulin from porcine brain ; Proc Natl Acad Sci U S A. 1981 Jul;78(7):4156-60.
- ↑ (en)Olivieri G, Brack C, Muller-Spahn F, Stahelin HB, Herrmann M, Renard P, Brockhaus M, Hock C. Mercury induces cell cytotoxicity and oxidative stress and increases beta-amyloid secretion and tau phosphorylation in SHSY5Y neuroblastoma cells. ; J Neurochem. 2000 Jan;74(1):231-6.
- ↑ (en)Olivieri G, Brack C, Muller-Spahn F, Stahelin HB, Herrmann M, Renard P, Brockhaus M, Hock C. ; Mercury induces cell cytotoxicity and oxidative stress and increases beta-amyloid secretion and tau phosphorylation in SHSY5Y neuroblastoma cells. ; J Neurochem. 2000 Jan;74(1):231-6.
- ↑ (en)Duhr EF, Pendergrass JC, Slevin JT, Haley BE.; HgEDTA complex inhibits GTP interactions with the E-site of brain beta-tubulin. Toxicol Appl Pharmacol. 1993 Oct;122(2):273-80.
- ↑ (en)Lorscheider FL, Vimy MJ, Summers AO, Zwiers H. ; The dental amalgam mercury controversy--inorganic mercury and the CNS; genetic linkage of mercury and antibiotic resistances in intestinal bacteria. ; Toxicology. 1995 Mar 31;97(1-3):19-22.
- ↑ (en)Leong CC, Syed NI, Lorscheider FL, Retrograde degeneration of neurite membrane structural integrity of nerve growth conesfollowing in vitro exposure to mercury. ; Neuroreport. 2001 Mar 26;12(4):733-7.
- ↑ (en)Cedrola S, Guzzi G, Ferrari D, Gritti A, Vescovi AL, Pendergrass JC, La Porta CA, Inorganic mercury changes the fate of murine CNS stem cells. ; The FASEB Journal mai 2003 ; 17(8):869-71. Epub 2003 Mar 28.
- ↑ (en)Fonte J, Miklossy J, Atwood C, Martins R, The severity of cortical Alzheimer's type changes is positively correlated with increased amyloid-beta Levels: Resolubilization of amyloid-beta with transition metal ion chelators. ; J Alzheimers Dis. avril 2001 ; 3(2):209-219.
- ↑ (en)Bush AI. ; Metal complexing agents as therapies for Alzheimer's disease ; Neurobiol Aging. novembre -décembre 2002 ; 23(6):1031-8.
- ↑ (en)Danbolt NC ; Glutamate uptake. ; Prog Neurobiol. septembre 2001 ; 65(1):1-105.
- ↑ (en)Brookes N. ; In vitro evidence for the role of glutamate in the CNS toxicity of mercury. ; Toxicology. 1992 Dec 4;76(3):245-56.
- ↑ (en)Mutkus L, Aschner JL, Syversen T, Shanker G, Sonnewald U, Aschner M. ; Mercuric chloride inhibits the in vitro uptake of glutamate in GLAST- and GLT-1-transfected mutant CHO-K1 cells.
- ↑ (en)Allen JW, Mutkus LA, Aschner M ; Mercuric chloride, but not methylmercury, inhibits glutamine synthetase activity in primary cultures of cortical astrocytes ; Brain Res. février 2001 9;891(1-2):148-57.
- ↑ (en)Boyd E. Haley (Université du Kentucky) ; The Relationship of the toxic effects of mercury to exacerbation of the medical condition classified as Alzheimer’s disease (2002) ; (étude accessible sur le site de l’UNEP).
- ↑ (en)Aksenov M, Aksenova M, Butterfield DA, Markesbery WR. ; Oxidative modification of creatine kinase BB in Alzheimer's disease brain. ; J Neurochem. juin 2000;74(6):2520-7.
- ↑ (en)David S, Shoemaker M, Haley BE. ; Abnormal properties of creatine kinase in Alzheimer's disease brain: correlation of reduced enzyme activity and active site photolabeling with aberrant cytosol-membrane partitioning ; Brain Res Mol Brain Res. mars 1998 ; 1;54(2):276-87.
- ↑ Selon le rapport du prof. Ménard.
- ↑ Étude Pixel[PDF].
- ↑ HAS : Maladie d'Alzheimer et maladies apparentées : suivi médical des aidants naturels.
- ↑ (en)Wilson RS, The relation of cognitive activity to risk of developing Alzheimer's disease. Neurology, 27 juin 2007.
- ↑ Faire des études préserverait de la maladie d'Alzheimer sur tempsreel.nouvelobs.com, 2 août 2010.
- ↑ (en)Carol Brayne, Paul G. Ince, Hannah A. D. Keage1, Ian G. McKeith, Fiona E. Matthews, Tuomo Polvikoski et Raimo Sulkava, Education, the brain and dementia: neuroprotection or compensation?, 6 mai 2010, Oxford Journals, Brain, Volume 133, Issue 8, p. 2210-2216 version intégrale.
- ↑ (en) Lautenschlager NT, Cox KL, Flicker L, Foster JK, van Bockxmeer FM, Xiao J, Greenop KR, Almeida OP. « Effect of physical activity on cognitive function in older adults at risk for Alzheimer disease: a randomized trial » JAMA 2008;300(9):1027-37. PMID 18768414 DOI:10.1001/jama.300.9.1027 Erratum in JAMA 2009 Jan 21;301(3):276.
- ↑ (en) Luchsinger JA, Reitz C, Honig LS, Tang MX, Shea S, Mayeux R. « Aggregation of vascular risk factors and risk of incident Alzheimer disease », Neurology 2005;65:545-51 PMID 16116114.
- ↑ (en)Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol 2000;57:1439-43.(résumé).
- ↑ A comparative study on the effect of high cholesterol diet on the hippocampal CA1 area of adult and aged rats
- ↑ Effects of a Saturated Fat and High Cholesterol Diet on Memory and Hippocampal Morphology in the Middle-Aged Rat
- ↑ (en) Methionine in Meat, Other Foods May Increase Alzheimer's Risk, sur le site emaxhealth.com du 17 décembre 2009.
- ↑ (en) Too much of red meat, fish may cause Alzheimer’s, sur le site indiatimes.com du 17 décembre 2009.
- 1 2 ALZHEIMER ET LA SANTÉ MENTALE, sur le site massacreanimal.org.
- ↑ (en) Alzheimer's and Brain Health, sur le site peta.org.
- ↑ (en) Newsletter of the Young Indian Vegetarians[PDF], sur le site youngindianvegetarians.co.uk.
- ↑ (en)Too much of red meat fish garlic yogurt may raise Alzheimers risk, sur le site expressindia.com.
- ↑ (en) Albanese E, Dangour AD, Uauy R, Acosta D, Guerra M, Guerra SS, Huang Y, Jacob KS, de Rodriguez JL, Noriega LH, Salas A, Sosa AL, Sousa RM, Williams J, Ferri CP, Prince MJ. « Dietary fish and meat intake and dementia in Latin America, China, and India: a 10/66 Dementia Research Group population-based study », Am J Clin Nutr. 2009;90(2):392-400. PMID 19553298 DOI:10.3945/ajcn.2009.27580.
- ↑ Food combination and Alzheimer disease risk: a protective diet
- ↑ Dietary intake of fish and omega-3 fatty acids in relation to long-term dementia risk
- ↑ Prevalence of dementia in older Japanese-American men in Hawaii: The Honolulu-Asia Aging Study
- ↑ Incidence of dementia and Alzheimer disease in 2 communities: Yoruba residing in Ibadan, Nigeria, and African Americans residing in Indianapolis, Indiana.
- 1 2 Dietary fat intake and the risk of incident dementia in the Rotterdam Study
- ↑ Dietary intakes of vitamin E, vitamin C, and β-carotene and risk of Alzheimer's disease: a meta-analysis
- 1 2 Dietary intake and cognitive function in a group of elderly people
- ↑ Dietary fat intake and 6-year cognitive change in an older biracial community population
- ↑ (en)Qiu C, Winblad B, Fratiglioni L. The age-dependent relation of blood pressure to cognitive function and dementia. Lancet Neurol 2005;4:487-99 (« CrossRef » (Archive • Wikiwix • Archive.is • Google • Que faire ?), consulté le 8 avril 2013).
- ↑ Nutrition and Alzheimer's disease: The detrimental role of a high carbohydrate diet[PDF].
- ↑ Alzheimer's Disease Is Type 3 Diabetes–Evidence Reviewed
- ↑ Diet Affects Markers of Alzheimer's Disease
- ↑ Reversals of age-related declines in neuronal signal transduction, cognitive, and motor behavioral deficits with blueberry, spinach, or strawberry dietary supplementation
- ↑ (en) Indian village may hold key to beating dementia.
- ↑ (en) http://healthmad.com/conditions-and-diseases/beware-of-meat-consumption
- ↑ Mediterranean diet, cognitive function, and dementia: a systematic review
- ↑ Western pattern diet raises Alzheimer’s risk
- ↑ (en)Fish Oil Boosts Brain Power.
- ↑ (en)The effects of omega-3 fatty acids monotherapy in Alzheimer's disease and mild cognitive impairment: A preliminary randomized double-blind placebo-controlled study.
- ↑ (en) Arendash GW, Cao C. « Caffeine and Coffee as Therapeutics Against Alzheimer's Disease », J Alzheimers Dis. 2010;20 Suppl 1:S117-26. PMID 20182037 DOI:10.3233/JAD-2010-091249.
- ↑ http://www.neurobiologyofaging.org/article/S0197-4580%2814%2900284-X/abstract
- ↑ Selon les travaux de recherche présentés lors du 4e Symposium scientifique international sur le thé et la santé humaine, Washington D.C. ; janvier 2008.
- ↑ Le thé qui soigne, article du Journal de Montréal (16 janvier 2008).
- ↑ Saint Louis University Animal Research Reinforces Conventional Wisdom: EVOO Is Good for You
- ↑ Serum lycopene, lutein and zeaxanthin, and the risk of Alzheimer's disease mortality in older adults
- ↑ Astaxanthin prevents heart disease, diabetes and Alzheimer's dementia
- ↑ The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse
- ↑ Alzheimer : un régime qui réduit le risque jusqu'à 53%
- ↑ MIND diet associated with reduced incidence of Alzheimer's disease
- ↑ Reversal of cognitive decline: A novel therapeutic program
- ↑ Fasting can help protect against brain diseases, scientists say
- ↑ Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer's disease
- ↑ Meal skipping helps rodents resist diabetes, brain damage
- ↑ (en)Riemersma-van der Lek et coll. ; Effect of bright light and melatonin on cognitive and non cognitive function of elderly residents of group care facilities. A randomized controlled trial. Revue JAMA 2008 ; 299 : 2642-2655.
- ↑ Cité dans un Article de Julie Luong intitulé Lumière et mélatonine contre la maladie d'Alzheimer, sur info-alzheimer.be (15 octobre 2008).
- ↑ (en)Kehoe PG, Wilcock GK. Is inhibition of the renin-angiotensin system a new treatment option for Alzheimer’s disease? ; Lancet Neurol 2007;6:373-8.
- ↑ (en)Skoog I, Lithell H, Hansson L, Elmfeldt D, Hofman A, Olofsson B, et al. ; Effect of baseline cognitive function and antihypertensive treatment on cognitive and cardiovascular outcomes: Study on COgnition and Prognosis in the Elderly (SCOPE). Am J Hypertens 2005;18:1052-9. (CrossRef).
- ↑ (en)Forette F, Seux ML, Staessen JA, Thijs L, Babarskiene MR, Babeanu S, et al. The prevention of dementia with antihypertensive treatment: new evidence from the Systolic Hypertension in Europe (Syst-Eur) study. Arch Intern Med 2002;162:2046-52. (Résumé).
- ↑ (en)Ara S. Khachaturian et al. ; Antihypertensive Medication Use and Incident Alzheimer Disease ; Arch Neurol. 2006;63:686-692. Mis en lgine le 13 mars 2006 (doi:10.1001/archneur.63.5.noc60013) (Résumé).
- ↑ (en)Matchar DB, McCrory DC, Orlando LA, Patel MR, Patel UD, Patwardhan MB, et al. ; Systematic review: comparative effectiveness of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers for treating essential hypertension. ; Ann Intern Med 2008;148:16-29.
- ↑ (en)Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet 2000;356:1627-31.
- ↑ Anti-Inflammatory Drugs and Risk of Alzheimer's Disease: An Updated Systematic Review and Meta-Analysis
- ↑ Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer's disease: systematic review and meta-analysis of observational studies
- ↑ - Un malade d'Alzheimer sur deux ne serait pas pris en charge, Sciences et Avenir, publié le 8 mai 2015
- ↑ Un malade d’Alzheimer sur deux ne serait pas diagnostiqué en France, Le Monde, 5 mai 2015
- 1 2 Polydor J-P, Alzheimer, mode d'emploi, le livre des aidants, préfacé par Madeleine Chapsal, prix Femina, L'esprit du temps Édition, 2009 (ISBN 2847951717).
- ↑ Laroche ML et al., Médicaments potentiellement inappropriés aux personnes âgées : intérêt d’une liste adaptée à la pratique médicale française. Rev Med Int 2009.
- 1 2 3 Haute Autorité de Santé « Maladie d’Alzheimer et maladies apparentées : prise en charge des troubles du comportement perturbateurs », argumentaire, HAS, mai 2009.
- ↑ Ricour C., Brutsaert S., Boland B., « Psychotropes au lieu de vie des personnes âgées, avec ou sans démence », Poster, Les rencontres Prescrire 2010.
- ↑ Benoit M, Staccini P, Brocker P, Benhamidat T, Bertogliati C, Lechowski L, et al. Symptômes comportementaux et psychologiques dans la maladie d'Alzheimer : résultats de l'étude REAL.FR. Rev Med Interne 2003;24(Suppl 3):319s-24s.
- ↑ (en)Aalten P, Verhey FR, Boziki M, Bullock R, Byrne EJ, Camus V, et al. ; Neuropsychiatric syndromes in dementia. Results from the European Alzheimer Disease Consortium: part I. Dement Geriatr Cogn Disord 2007;24(6):457-63.
- ↑ (en)Banerjee S, Hellier J, Dewey M, et al. Sertraline or mirtazapine for depression in dementia (HTA-SADD): a randomised, multicentre, double-blind, placebo-controlled trial, Lancet, 2011;378:403-411.
- ↑ Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA 2005;293(5):596-608.
- ↑ (en)Belmin J, Péquignot R, Konrat C, Pariel-Madjlessi S. Management of Alzheimer disease, Presse Med. 2007 Oct;36(10 Pt 2):1500-10. Epub 2007 Jun 29.
- ↑ (en)Rochon PA, Normand SL, Gomes T, et al. ; Antipsychotic therapy and short-term serious events in older adults with dementia. Arch Intern Med 2008;168:1090-6.
- ↑ Chevalier P., Continuer ou arrêter les neuroleptiques chez des patients déments ? Minerva 2009 8(1); 11-11.
- ↑ (en)Curran HV, Collins R, Fletcher S, Kee SC, Woods B, Iliffe S. Older adults and withdrawal from benzodiazepine hypnotics in general practice: effects on cognitive function, sleep, mood and quality of life. Psychol Med. 2003 Oct;33(7):1223-37.
- ↑ (en)Courtney C, Farrell D, Gray R, Hills R, Lynch L, Sellwood E, Edwards S, Hardyman W, Raftery J, Crome P, Lendon C, Shaw H, Bentham P; AD2000 Collaborative Group. Long-term donepezil treatment in 565 patients with Alzheimer's disease (AD2000): randomised double-blind trial. Lancet 2004;363:2105-15. PMID 15220031.
- ↑ Amélioration du service médical rendu.
- ↑ la revue Prescrire, no 293, mars 2008.
- ↑ (en) (National Institute for Health and Clinical Excellence): Alzheimer's disease - donepezil, galantamine, rivastigmine (review) and memantine, sur guidance.nice.org.uk.
- ↑ Revue Prescrire, Numéro 278, Médicaments de la maladie d'Alzheimer : à l'origine de tremblements et d'aggravation de symptômes parkinsoniens, décembre 2006.
- ↑ Revue Prescrire ; Les médicaments de la maladie d'Alzheimer, Mini dossier mis à jour le 15 février 2007.
- ↑ (en) Donepezil, galantamine, rivastigmine (review) and memantine for the treatment of Alzheimer's disease, National Institute for Health and Clinical Excellence (NICE).
- ↑ Réévaluation des médicaments Alzheimer, sur le site has-sante.fr.
- ↑ (en)Areosa SA, McShane R, Sherriff F. Cochrane Database Syst Rev 2004(4);CD003154.pub2. PMID Memantine for dementia., sur le site nlm.nih.gov.
- ↑ .
- ↑ D'après actualites-news-environnement.com.
- ↑ (en)Holmes C, Boche D, Wilkinson D, et al. Long-term effects of Aβ42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial, Lancet 2008;372:216-223.
- ↑ Le Pr Tohru Hasegawa travaille au Women's Junior College de Saga au Japon.
- ↑ (en)HASEGAWA Tohru, MIKODA Nobuyuki, KITAZAWA Masashi, LAFERLA Frank M. « Treatment of Alzheimer's Disease with Anti-Homocysteic Acid Antibody in 3xTg-AD Male Mice », PLoS ONE 5(1): e8593 - 20/01/2010 - doi:10.1371/journal.pone.0008593.
- ↑ Japon numéro 528 (1/02/2010) - Ambassade de France au Japon / ADIT.
- ↑ (en)Arendash GW, Sanchez-Ramos J, Mori T, Mamcarz M, Lin X, Runfeldt M, Wang L, Zhang G, Sava V, Tan J, Cao C. ; Electromagnetic field treatment protects against and reverses cognitive impairment in Alzheimer's disease mice. ; J Alzheimers Dis. 2010 ; Jan;19(1):191-210. PMID 20061638.
- ↑ « Extraire le cholestérol du cerveau pour combattre Alzheimer », sur www.sciencesetavenir.fr (consulté le 4 octobre 2015)
- 1 2 3 (en)World Alzheimer Report 2010[PDF].
- ↑ Source : Alzheimer's Disease International.
- ↑ Société Alzheimer Canada: Facteurs de risque
- ↑ (en) Kuller LH. « Dementia epidemiology research: it is time to modify the focus of research » J Gerontol A Biol Sci Med Sci. 2006;61(12):1314-8. PMID 17234827
- ↑ Alzheimer’s Epidemic Worse In Some Regions
- ↑ World Health Rankings ALZHEIMERS/DEMENTIA
- ↑ CDC: Mortality From Alzheimer's Disease in the United States: Data for 2000 and 2010
- ↑ (en) Chandra V, Pandav R, Dodge HH, Johnston JM, Belle SH, DeKosky ST, Ganguli M. « Incidence of Alzheimer's disease in a rural community in India: the Indo-US study » Neurology 2001;57(6):985-9. PMID 11571321
- ↑ (en)Hendrie HC, Hall KS, Ogunniyi A, Gao S ; Alzheimer's disease, genes, and environment: the value of international studies. ; Can J Psychiatry. 2004 Feb;49(2):92-9.
- ↑ (en)Hendrie HC ; Lessons learned from international comparative crosscultural studies on dementia, Am J Geriatr Psychiatry, juin 2006 ; 14(6):480-8.
- 1 2 (en)Shadlen MF, Larson EB, Yukawa M. The epidemiology of Alzheimer's disease and vascular dementia in Japanese and African-American populations: the search for etiological clues. ; Department of Medicine, Harborview Medical Center, School of Pharmacy, University of Washington, Seattle, États-Unis. Neurobiol Aging. 21 mars 2000 ; (2):171-81.
- ↑ (en)Graves AB, Larson EB, Edland SD, Bowen JD, McCormick WC, McCurry SM, Rice MM, Wenzlow A, Uomoto JM. Prevalence of dementia and its subtypes in the Japanese American population of King County, Washington state. The Kame Project. Department of Epidemiology and Biostatistics, University of South Florida, Tampa, États-Unis. Am J Epidemiol. 1996 octobre 15;144(8):760-71.
- ↑ (en)Yamada T, Kadekaru H, Matsumoto S, Inada H, Tanabe M, Moriguchi EH, Moriguchi Y, Ishikawa P, Ishikawa AG, Taira K, Yamori Y. ; Prevalence of dementia in the older Japanese-Brazilian population. ; Psychiatry Clin Neurosci. 2002 Feb;56(1):71-5 (Department of Internal Medicine and Health Care, Fukuoka University, Fukuoka, Japan).
- ↑ (en)Lopes MA, Bottino CM. ; Prevalence of dementia in several regions of the world: analysis of epidemiologic studies from 1994 to 2000 (Instituto de Psiquiatria, Hospital das Clínicas, Faculdade de Medicina, Universidade de São Paulo, Brasil). Arq Neuropsiquiatr. mars 2002 ;60(1):61-9.
- ↑ (en)Fujishima M, Kiyohara Y. ; Incidence and risk factors of dementia in a defined elderly Japanese population: the Hisayama study. (Department of Medicine and Clinical Sciences, Kyushu University, Japon) ; Ann N Y Acad Sci. ; novembre 2002; Nov;977:1-8.
- ↑ Rapport 2010 de l'Association Alzheimer américaine, figures 12 et 13[PDF].
- ↑ Nouvelles percées concernant la maladie d’Alzheimer avec des souris transgéniques[PDF] ; L'écho des souris, 3 septembre 2011.
- ↑ (en)Wancata J, Musalek M, Alexandrowicz R, Krautgartner ; Number of dementia sufferers in Europe between the years 2000 and 2050. M. Eur Psychiatry. 2003 Oct;18(6):306-13.
- ↑ « Taux de patients belges atteints de la maladie d'Alzheimer », sur Le centre hospitalier chrétien (CHC) (consulté le 14 janvier 2006).
- ↑ no 2454 - Rapport sur la maladie d'Alzheimer et les maladies apparentées au nom de l'Office parlementaire d'évaluation des politiques de santé(rapporteuse : Mme Cécile Gallez).
- ↑ (en)Ramaroson H, Helmer C, Barberger-Gateau P, Letenneur L, Dartigues JF ; Prevalence of dementia and Alzheimer's disease among subjects aged 75 years or over : Résultats mis à jour du programme PAQUID cohort] PAQUID. ; Unité INSERM 330, Université Victor Segalen Bordeaux II. Rev Neurol (Paris). 2003 Apr;159(4):405-11.
- ↑ (en) Helmer C., Pasquier F, Dartigues JF. Epidemiology of Alzheimer disease and related disorders Helmer Med Sci (Paris). 2006 Mar;22(3):288-96}.
- ↑ Auguste D. :
- (de) Alzheimer Alois, « Über eine eigenartige Erkrankung der Hirnrinde », Allgemeine Zeitschrift fur Psychiatrie und Psychisch-Gerichtlich Medizin, vol. 64, no 1–2, , p. 146–148
- (en) Alzheimer Alois, « About a peculiar disease of the cerebral cortex », Alzheimer Dis Assoc Disord, vol. 1, no 1, , p. 3–8 (PMID 3331112)
- (en) Maurer Ulrike, Maurer Konrad, Alzheimer: the life of a physician and the career of a disease, New York, Columbia University Press, (ISBN 0-231-11896-1), p. 270.
- ↑ (en) Berrios G E, « Alzheimer's disease: a conceptual history », Int. J. Ger. Psychiatry, vol. 5, no 6, , p. 355–365 (DOI 10.1002/gps.930050603).
- ↑ (en) Kraepelin Emil, Diefendorf A. Ross (traduit par), Clinical Psychiatry: A Textbook For Students And Physicians (Reprint), Kessinger Publishing, (ISBN 1-4325-0833-4), p. 568
- ↑ (en) Katzman Robert, Terry Robert D, Bick Katherine L (editors), Alzheimer's disease: senile dementia and related disorders, New York, Raven Press, (ISBN 0-89004-225-X), p. 595
- ↑ (en) Boller F, Forbes MM, « History of dementia and dementia in history: an overview », J Neurol Sci, vol. 158, no 2, , p. 125–133. (PMID 9702682, DOI 10.1016/S0022-510X(98)00128-2)
- ↑ (en) Amaducci LA, Rocca WA, Schoenberg BS, « Origin of the distinction between Alzheimer's disease and senile dementia: how history can clarify nosology », Neurology, vol. 36, no 11, , p. 1497–1499. (PMID 3531918)
- ↑ [PDF] Document de 140 pages sur l’observatoire national sur la recherche sur la maladie d'Alzheimer, voir aussi le site de l'ONRA.
- ↑ Dr Douçot-Hermelin (Revue de Gériatrie 2008;33:787.
Annexes
Articles connexes
Liens externes
Catégorie Alzheimer de l’annuaire DMOZ
- Page du Gouvernement français concernant la maladie d'Alzheimer
- Portail d'Alzheimer de l'Union européenne partiellement en français
- Société Alzheimer du Canada
- Portail gouvernemental du Plan Alzheimer
- (en) Journal of Alzheimer's Disease
- (en) Journal of Alzheimer's & Dementia
Guides et recommandations
- diagnostic et la prise en charge de la maladie d’Alzheimer et apparentées a été publiée le 16 décembre 2011 par la Haute Autorité de santé.*
- RSI, IRCEM et France Alzheimer (2014) Guide interactif pour permettre aux indépendants de mieux connaître la maladie et mieux accompagner les personnes atteintes.
 Portail de la médecine
Portail de la médecine  Portail des neurosciences
Portail des neurosciences  Portail de la psychologie
Portail de la psychologie  Portail du handicap
Portail du handicap