
{kind=link}

Destilaci??n
Antecedentes
Esta selecci??n se hace para las escuelas por caridad para ni??os leer m??s . Una buena manera de ayudar a otros ni??os es mediante el patrocinio de un ni??o
La destilaci??n es un m??todo de separar sustancias qu??micas basadas en diferencias en su volatilidades en una mezcla l??quida hirviendo. Destilaci??n por lo general forma parte de un proceso qu??mico m??s grande, y se refiere as?? a como una funcionamiento de la unidad.
Comercialmente, la destilaci??n tiene un n??mero de usos. Se utiliza para separar el petr??leo crudo en m??s fracciones para usos espec??ficos, tales como el transporte , la generaci??n de electricidad y calefacci??n. El agua se destil?? para eliminar impurezas, tales como la sal del agua del mar. El aire se destila para separar sus componentes - en particular de ox??geno , nitr??geno y arg??n - para el uso industrial. Destilaci??n de fermentados soluciones se ha utilizado desde la antig??edad para producir bebidas destiladas con un contenido de alcohol superior.
Historia
Las primeras formas de destilaci??n se sab??a que babil??nicos alquimistas en Mesopotamia (en lo que hoy es Irak ) de al menos el Segundo milenio antes de Cristo. La destilaci??n fue luego conocido por griegos alquimistas del siglo 1 dC, y el posterior desarrollo de un aparato de destilaci??n a gran escala se produjo en respuesta a las demandas de los esp??ritus. Hypathia de Alejandr??a se le atribuye haber inventado un aparato de destilaci??n temprano, y la primera descripci??n exacta del aparato de destilaci??n est?? dada por Z??simo de Alejandr??a en el siglo IV.

En el siglo octavo, los alquimistas en el Medio Oriente producen procesos de destilaci??n para purificar sustancias qu??micas para la industria fines tales como el aislamiento natural, ??steres ( perfumes) y la producci??n de puro alcohol . El primero de ellos fue el persa Jabir Ibn Hayyan (Geber) alrededor del a??o 800 dC, a quien se atribuye la invenci??n de aparatos qu??micos numerosos y procesos que todav??a est??n en uso hoy en d??a. En particular, su alambique fue el primero a??n con retortas que podr??a purificar completamente los productos qu??micos, un precursor de la alambique, y su dise??o ha servido como inspiraci??n para un aparato de destilaci??n de micro-escala moderna como la stillhead Hickman. Petr??leo se destil?? por primera vez por otro persa , al-Razi (Rhazes) en el siglo noveno, para producir queroseno, mientras destilaci??n de vapor fue inventada por Avicena a principios del siglo 11, para la producci??n de aceites esenciales . Como la alquimia se desarroll?? en la ciencia de la qu??mica , los vasos llamados retortas se acostumbraron para destilaciones. Ambos alambiques y retortas son formas de cristaler??a con largos cuellos que apuntan hacia un lado en un ??ngulo descendente que actuaban como aire fr??o- condensadores a condensar el destilado y se deja gotear hacia abajo para la colecci??n.
M??s tarde, se inventaron alambiques de cobre. Uniones remachadas a menudo se mantienen apretadas utilizando diversas mezclas, por ejemplo, una masa hecha de harina de centeno. Estos alambiques ofrecen a menudo un sistema de refrigeraci??n alrededor del pico, usando agua fr??a, por ejemplo, lo que hizo que la condensaci??n de alcohol m??s eficiente. Estos fueron llamados alambiques.
Hoy en d??a, las retortas y alambiques de haber sido suplantado en gran parte por m??todos de destilaci??n m??s eficientes en la mayor??a de los procesos industriales. Sin embargo, el bote sigue siendo ampliamente utilizado para la elaboraci??n de algunos alcoholes finos como co??ac, El whisky escoc??s y algunos vodkas. Alambiques de diferentes materiales (madera, arcilla, acero inoxidable) tambi??n son utilizados por contrabandistas en varios pa??ses. Los peque??os alambiques tambi??n se venden para la producci??n nacional de agua de flores o aceites esenciales .
Aplicaciones de la destilaci??n
La aplicaci??n de la destilaci??n pueden dividirse en cuatro grupos: a escala de laboratorio , de destilaci??n industrial , destilaci??n de hierbas para perfumer??a y productos medicinales ( destilado de hierbas) y el procesamiento de alimentos . Los dos ??ltimos son distintos de los dos anteriores, en que en la destilaci??n no se utiliza como un m??todo de purificaci??n cierto, pero m??s para transferir todos vol??tiles de los materiales de base para el destilado.
La principal diferencia entre la destilaci??n escala de laboratorio y la destilaci??n industrial es que la destilaci??n escala de laboratorio se realiza a menudo en forma discontinua, mientras que la destilaci??n industrial a menudo se produce continuamente. En la destilaci??n por lotes, la composici??n del material de origen, los vapores de los compuestos de destilaci??n y el cambio destilado durante la destilaci??n. En la destilaci??n por lotes, una todav??a est?? cargada (suministrado) con un lote de mezcla de alimentaci??n, que se separa en sus fracciones de componentes que se recogen secuencialmente de m??s vol??til a menos vol??til, con los fondos restante de la fracci??n (menos o no vol??til) retirados al final. El todav??a puede entonces ser recargada y el proceso se repite.
En destilaci??n continua, los materiales de base, los vapores y el destilado se mantienen a una composici??n constante mediante la reposici??n cuidadosamente el material de origen y la eliminaci??n de las fracciones tanto de vapor y l??quido en el sistema. Esto resulta en un mejor control del proceso de separaci??n.
Modelo destilaci??n idealizada
El punto de ebullici??n de un l??quido es la temperatura a la que la la presi??n de vapor del l??quido es igual a la presi??n que rodea el l??quido. El punto de ebullici??n normal de un l??quido es el caso especial en que la presi??n de vapor del l??quido es igual a la ambiente presi??n atmosf??rica. Un l??quido en un recipiente a una presi??n inferior a la presi??n atmosf??rica hierve a temperatura m??s baja que el punto de ebullici??n normal, y un l??quido en un recipiente a una presi??n mayor que la presi??n atmosf??rica hervir?? a una temperatura mayor que el punto de ebullici??n normal. En otras palabras, todos los l??quidos tienen un n??mero infinito de puntos de ebullici??n.
Es un error muy com??n que en una mezcla l??quida a una presi??n dada, cada componente hierve a la temperatura de ebullici??n correspondiente a la presi??n dada y los vapores de cada componente recoger?? por separado y puramente. Esto, sin embargo, no se produce incluso en un sistema idealizado. Modelos idealizados de destilaci??n se rigen esencialmente por La ley de Raoult y La ley de Dalton.
La ley de Raoult asume que un componente contribuye al total presi??n de vapor de la mezcla en proporci??n a su porcentaje de la mezcla y su presi??n de vapor en estado puro. Si un componente cambia la presi??n de vapor del otro componente, o si la volatilidad de un componente depende de su porcentaje en la mezcla, la ley fallar??.
La ley de Dalton establece que la presi??n de vapor total es la suma de las presiones de vapor de cada componente individual en la mezcla. Cuando se calienta un l??quido multi-componente, la presi??n de vapor de cada componente se elevar??, causando as?? la presi??n de vapor total en aumento. Cuando la presi??n de vapor total alcanza la presi??n que rodea el l??quido, de ebullici??n se produce y el l??quido se convierte en gas a lo largo de la mayor parte del l??quido. Tenga en cuenta que una mezcla dada tiene un punto de ebullici??n a una presi??n dada, cuando los componentes son mutuamente soluble.
El modelo idealizado es exacta en el caso de l??quidos qu??micamente similares, tales como benceno y tolueno . En otros casos, se observan desviaciones graves de la ley de Raoult y la ley de Dalton, el m??s famoso en la mezcla de etanol y agua. Estos compuestos, cuando se calientan juntos, forman una aze??tropo, en el que la temperatura de ebullici??n de la mezcla es inferior a la temperatura de ebullici??n de cada l??quido separado. Virtualmente todos los l??quidos, cuando se mezcla y se calienta, se mostrar?? el comportamiento azeotr??pica. Aunque hay m??todos computacionales que se pueden utilizar para estimar el comportamiento de una mezcla de componentes arbitrarios, la ??nica manera de obtener informaci??n precisa los datos de equilibrio vapor-l??quido es por medici??n.
No es posible purificar completamente una mezcla de componentes por destilaci??n, ya que esto requerir??a cada componente en la mezcla para tener un cero presi??n parcial. Si los productos ultra-puros son la meta, luego m??s separaci??n qu??mica debe ser aplicada.
Destilaci??n por lotes
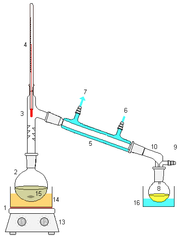
El calentamiento de una mezcla ideal de dos sustancias vol??tiles A y B (con A que tiene la mayor volatilidad, o menor punto de ebullici??n) en una configuraci??n de destilaci??n por lotes (tal como en un aparato representado en la figura apertura) hasta que la mezcla est?? hirviendo resultados en un vapor por encima del l??quido que contiene una mezcla de A y B. La relaci??n entre A y B en el vapor ser?? diferente de la relaci??n en el l??quido: la relaci??n en el l??quido ser?? determinado por c??mo se prepar?? la mezcla original, mientras que el ratio en el vapor ser?? enriquecida en el compuesto m??s vol??til, A (debido a la ley de Raoult, v??ase m??s arriba). El vapor pasa a trav??s del condensador y se elimina del sistema. Esto a su vez significa que la relaci??n de los compuestos en el l??quido restante es ahora diferente de la relaci??n inicial (es decir, m??s enriquecida en B que el l??quido de partida).
El resultado es que la relaci??n en la mezcla l??quida est?? cambiando, convirti??ndose m??s rico en el componente B. Esto hace que el punto de ebullici??n de la mezcla a aumentar, lo que a su vez se traduce en un aumento de la temperatura en el vapor, lo que resulta en un cambio de la relaci??n de A: B en la fase gas (como destilaci??n contin??a, hay una creciente proporci??n de B en la fase de gas). Esto resulta en un cambio de la relaci??n lentamente A: B en el destilado.
Si la diferencia de presi??n de vapor entre los dos componentes A y B es grande (generalmente expresada como la diferencia en los puntos de ebullici??n), la mezcla en el comienzo de la destilaci??n es altamente enriquecido en el componente A, y cuando el componente A ha destilaci??n, el l??quido hirviendo se enriquece en el componente B.
La destilaci??n continua
La destilaci??n continua es una destilaci??n continua en la que una mezcla l??quida es continua (sin interrupci??n) introducen en el proceso y las fracciones separadas se retiran continuamente como flujos de salida seg??n pasa el tiempo durante la operaci??n. La destilaci??n continua produce al menos dos fracciones de salida, incluyendo al menos una fracci??n de destilado vol??til, que ha sido hervida y capturado por separado como un vapor condensado a un l??quido. Siempre hay un fondo (o residuo) fracci??n, que es el residuo menos vol??til que no ha sido capturado por separado como un vapor condensado.
Mejoras generales
Ambos destilaciones por lotes y continuos se pueden mejorar mediante el uso de una columna de fraccionamiento en la parte superior del matraz de destilaci??n. La columna de separaci??n mejora al proporcionar un ??rea de superficie m??s grande para el vapor y condensado a entrar en contacto. Esto ayuda a que permanezca en equilibrio durante tanto tiempo como sea posible. La columna puede consistir incluso en peque??os subsistemas ('bandejas' o 'platos') que contienen todos una, mezcla l??quida hirviendo enriquecido, todas con su propio equilibrio l??quido-vapor.
Hay diferencias entre a escala de laboratorio y columnas de fraccionamiento a escala industrial, pero los principios son los mismos. Ejemplos de columnas de fraccionamiento a escala de laboratorio (en el aumento de la eficacia) incluyen:
- Condensador del aire
- Columna Vigreux (por lo general a escala de laboratorio solamente)
- Columna de relleno (lleno de perlas de vidrio, pedazos de metal u otro material qu??micamente inerte)
- Spinning sistema de destilaci??n banda
Laboratorio destilaci??n escala
Destilaciones escala de laboratorio se ejecutan casi exclusivamente como destilaciones por lotes. El dispositivo utilizado en la destilaci??n, se refiere a veces como una todav??a, consiste en un m??nimo de un rehervidor o bote en el que el material fuente se calienta, un condensador en el que se calienta el vapor se enfr??a de nuevo al l??quido de estado , y un receptor en el que el l??quido concentrado o purificado, llamado el destilado, es recogido. Varios t??cnicas a escala de laboratorio para la destilaci??n existe (v??ase tambi??n tipos de destilaci??n).
Destilaci??n simple
En la destilaci??n simple, todos los vapores calientes producidos se canalizan inmediatamente en un condensador que enfr??a y condensa los vapores. Por lo tanto, el destilado no ser?? puro - su composici??n ser?? id??ntica a la composici??n de los vapores a la temperatura y presi??n dadas, y puede ser calculado a partir de La ley de Raoult.
Como resultado, la destilaci??n simple se usa por lo general s??lo a los l??quidos separados cuyos puntos de ebullici??n difieren en gran medida (regla de oro es de 25 ?? C), o para separar l??quidos de s??lidos no vol??tiles o aceites. Para estos casos, las presiones de vapor de los componentes suelen ser suficientemente diferente que la ley de Raoult puede despreciarse debido a la escasa contribuci??n del componente menos vol??til. En este caso, el destilado puede ser suficientemente puro para su prop??sito previsto.
Destilaci??n fraccionada
Para muchos casos, los puntos de ebullici??n de los componentes en la mezcla ser??n lo suficientemente cerca para que la ley de Raoult debe tenerse en consideraci??n. Por lo tanto, la destilaci??n fraccionada se debe utilizar con el fin de separar los componentes bien por los ciclos de vaporizaci??n-condensaci??n repetidas dentro de una columna de fraccionamiento para llevar.
Como la soluci??n a ser purificado se calienta, sus vapores suben a la columna de fraccionamiento. A medida que asciende, se enfr??a, se condense en las paredes de condensador y las superficies del material de embalaje. Aqu??, el condensado contin??a para ser calentado por el aumento de los vapores calientes; se vaporiza una vez m??s. Sin embargo, la composici??n de los vapores frescas se determinan una vez m??s por la ley de Raoult. Cada ciclo de vaporizaci??n-condensaci??n (llamado plato te??rico) dar?? lugar a una soluci??n m??s pura del componente m??s vol??til. En realidad, cada ciclo a una temperatura dada no se produce exactamente en la misma posici??n en la columna de fraccionamiento; plato te??rico es por lo tanto un concepto en lugar de una descripci??n exacta.
M??s platos te??ricos conducen a mejores separaciones. La girar sistema de destilaci??n de banda utiliza una banda de hilatura de Teflon o metal para obligar a los vapores ascendentes en estrecho contacto con el condensado descendente, aumentando el n??mero de platos te??ricos.
La destilaci??n de vapor
Como la destilaci??n al vac??o, la destilaci??n de vapor es un m??todo de destilaci??n de compuestos que son sensibles al calor. Este proceso implica el uso de vapor de burbujeo a trav??s de una mezcla calentada de la materia prima. Por la ley de Raoult, algunos de los compuesto objetivo se vaporizar?? (de acuerdo con su presi??n parcial). La mezcla de vapor se enfr??a y se condensa, produciendo generalmente una capa de aceite y una capa de agua.
La destilaci??n de vapor de varios hierbas y flores arom??ticas pueden dar lugar a dos productos; un aceite esencial , as?? como un acuosa destilado a base de hierbas. Los aceites esenciales se utilizan a menudo en perfumer??a y aromaterapia mientras que los destilados acuosos tienen muchas aplicaciones en aromaterapia, procesamiento de alimentos y protecci??n de la piel.

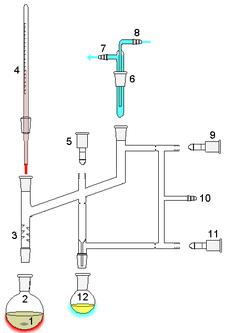
1: Agitador bar / perlas de ebullici??n 2: Todav??a olla 3: columna de fraccionamiento 4: Term??metro de temperatura / punto de ebullici??n 5: Teflon grifo 1 6: Cold dedo 7: El agua de refrigeraci??n a cabo 8: El agua de refrigeraci??n en 9: Teflon grifo 2 10: entrada de vac??o / gas 11: Teflon grifo 3 12: Todav??a receptor
La destilaci??n al vac??o
Algunos compuestos tienen puntos de ebullici??n muy altos. Para hervir tales compuestos, a menudo es mejor para bajar la presi??n a la que tales compuestos se hierven en lugar de aumentar la temperatura. Una vez que la presi??n baja a la presi??n de vapor del compuesto (a la temperatura dada), punto de ebullici??n y el resto del proceso de destilaci??n puede comenzar. Esta t??cnica se conoce como destilaci??n al vac??o y se encuentra com??nmente en el laboratorio en la forma de la evaporador rotatorio.
Esta t??cnica tambi??n es muy ??til para compuestos que hierven m??s all?? de su Por lo tanto, la temperatura de descomposici??n a presi??n atmosf??rica y que se descompone por cualquier intento de hervir a presi??n atmosf??rica.
La destilaci??n al vac??o de aire sensible
Algunos compuestos tienen altos puntos de ebullici??n as?? como ser sensible al aire. Un sistema simple de destilaci??n al vac??o como se ejemplifica anteriormente puede ser utilizado, por lo que el vac??o se sustituye con un gas inerte despu??s de la destilaci??n es completa. Sin embargo, este es un sistema menos satisfactoria si se desea recoger las fracciones bajo presi??n reducida. Para ello un adaptador "pig" se puede a??adir al extremo del condensador, o para obtener mejores resultados o para aire muy compuestos sensibles a una Aparato Perkin tri??ngulo se puede utilizar.
El tri??ngulo Perkin, tiene medios a trav??s de una serie de vidrio o Grifos de tefl??n para permite fracciones para ser aislados del resto de la Sin embargo, sin el cuerpo principal de la destilaci??n siendo retirado de ya sea la fuente de vac??o o calor, y por lo tanto puede permanecer en un estado de reflujo. Para ello, la muestra se aisl?? por primera vez desde el vac??o por medio de los grifos, el vac??o sobre la muestra se reemplaza entonces con un gas inerte (tal como nitr??geno o arg??n ) y luego se puede tap?? y se retira. Un recipiente de recogida fresco puede ser a??adido al sistema, se evacu?? y se vincula de nuevo en el sistema de destilaci??n a trav??s de los grifos para recoger una fracci??n de segundo, y as?? sucesivamente, hasta que se han recogido todas las fracciones.
Destilaci??n de corto recorrido

Destilaci??n de corto recorrido es una t??cnica de destilaci??n que implica el destilado de viajar una distancia corta, a menudo s??lo unos pocos cent??metros. Un ejemplo cl??sico ser??a una destilaci??n que implica el destilado que viaja de un bulbo de vidrio a otro, sin la necesidad de un condensador de separaci??n de las dos c??maras. Esta t??cnica se utiliza a menudo para los compuestos que son inestables a altas temperaturas. La ventaja es que la temperatura de calentamiento puede ser considerablemente inferior (en este presi??n reducida) que el punto de que el l??quido de ebullici??n a presi??n normal, y que el destilado s??lo tiene que recorrer una distancia corta antes de condensaci??n.
Otros tipos
- En evaporaci??n rotatoria un aparato de destilaci??n al vac??o se utiliza para eliminar a granel disolventes a partir de una muestra. T??picamente, el vac??o es generado por un agua aspirador o una bomba de membrana.
- En un Kugelrohr (> 300 ?? C) compuestos de un aparato de destilaci??n de corto recorrido se utiliza normalmente (por lo general en combinaci??n con un alto vac??o ()) para destilar alto punto de ebullici??n. El aparato consiste en un horno en el que se coloca el compuesto a destilada, una parte de recepci??n que est?? fuera del horno, y un medio de rotaci??n de la muestra. El vac??o se genera normalmente mediante el uso de una bomba de alto vac??o.
- El proceso de destilaci??n reactiva implica el uso del recipiente de reacci??n como sigue. En este proceso, el producto es por lo general significativamente menor punto de ebullici??n que sus reactivos. A medida que se forma el producto de los reactantes, se vaporiza y se separa de la mezcla de reacci??n. Esta t??cnica es un ejemplo de una continua vs. un proceso por lotes; ventajas incluyen menos tiempo de inactividad para cargar el recipiente de reacci??n con material de partida, y menos estudio diagn??stico.
- Destilaci??n destructiva implica el fuerte calentamiento de los s??lidos (material de frecuencia org??nico) en ausencia de ox??geno (para evitar la combusti??n) que se evapore varios l??quidos de alta ebullici??n, as?? como productos de term??lisis. Los gases desprendidos se enfr??an y se condensan como en la destilaci??n normal. La destilaci??n destructiva de la madera para dar metanol es la ra??z de su nombre com??n - alcohol de madera.
- La pervaporaci??n es un m??todo para la separaci??n de mezclas de l??quidos por vaporizaci??n parcial a trav??s de un no-porosa membrana.
- Destilaci??n en seco, a pesar de su nombre, no es verdaderamente destilaci??n, sino m??s bien una reacci??n qu??mica conocida como pir??lisis en la que las sustancias s??lidas se calientan en un fuertemente atm??sfera reductora y las fracciones vol??tiles son recogidos.
- La destilaci??n extractiva se define como la destilaci??n en presencia de un alto punto de ebullici??n miscible, componente relativamente no vol??til, el disolvente, que no forma aze??tropo con los otros componentes en la mezcla.
- La evaporaci??n instant??nea (o evaporaci??n parcial) es la vaporizaci??n parcial que se produce cuando una corriente de l??quido saturado se somete a una reducci??n de la presi??n pasando a trav??s de un estrangulamiento v??lvula u otro dispositivo de estrangulamiento. Este proceso es una de las operaciones unitarias simples.
- Destilaci??n Freeze es un m??todo an??logo de purificaci??n utilizando congelaci??n en lugar de la evaporaci??n. No es realmente destilaci??n, y no produce productos equivalentes a destilaci??n. Este proceso se utiliza en la producci??n de cerveza hielo y vino de hielo para aumentar el etanol y el az??car contenido, respectivamente.
- Destilaci??n conjunta es la destilaci??n que se lleva a cabo en mezclas en las que los dos compuestos no son miscibles.
La destilaci??n azeotr??pica
Las interacciones entre los componentes de la soluci??n crean propiedades ??nicas a la soluci??n, como la mayor??a de los procesos implican mezclas no ideales, donde La ley de Raoult no se sostiene. Tales interacciones pueden dar lugar a un punto de ebullici??n constante aze??tropo que se comporta como si se tratara de un compuesto puro (es decir, se reduce a una sola temperatura en lugar de un rango). En un aze??tropo, la soluci??n contiene el componente dado en la misma proporci??n que el vapor, por lo que la evaporaci??n no cambia la pureza, y la destilaci??n no afecta a la separaci??n. Por ejemplo, el alcohol et??lico y el agua forman un aze??tropo de 95% a 78,2 ?? C.
Si el aze??tropo no se considera suficientemente puro para su uso, existen algunas t??cnicas para romper el aze??tropo para dar un destilado puro. Este conjunto de t??cnicas se conoce como destilaci??n azeotr??pica. Algunas t??cnicas de ello por los "saltar" sobre la composici??n azeotr??pica (mediante la adici??n de un componente adicional para crear un nuevo aze??tropo, o mediante la variaci??n de la presi??n). Otros trabajan por qu??mica o f??sicamente eliminar o secuestrar la impureza. Por ejemplo, para purificar m??s all?? de etanol 95%, un agente de secado o una desecante tal como carbonato de potasio puede ser a??adido para convertir el agua soluble en insoluble agua de cristalizaci??n. Los tamices moleculares se utilizan a menudo para este prop??sito tambi??n.
L??quidos inmiscibles, tales como agua y tolueno, forma f??cilmente aze??tropos. Com??nmente, estos aze??tropos se conocen como un aze??tropo de bajo punto de ebullici??n debido a que el punto de ebullici??n del aze??tropo es menor que el punto de cualquiera de los componentes puros de ebullici??n. La temperatura y la composici??n del aze??tropo es f??cilmente predecir a partir de la presi??n de vapor de los componentes puros, sin el uso de la ley de Raoult. El aze??tropo se rompe f??cilmente en una destilaci??n configuraci??n mediante el uso de un separador de l??quido-l??quido (un decantador) para separar las dos capas l??quidas que se condensan por encima. S??lo una de las dos capas l??quidas se somete a reflujo a la destilaci??n set-up.
Tambi??n existen aze??tropos alto punto de ebullici??n, tales como una mezcla de 20 por ciento en peso de ??cido clorh??drico en agua. Como implica el nombre, el punto de ebullici??n del aze??tropo es mayor que el punto de cualquiera de los componentes puros de ebullici??n.
Para romper destilaciones azeotr??picas y los l??mites de destilaci??n transversales, tales como en el problema DeRosier, es necesario aumentar la composici??n de la clave de la luz en el destilado.
Fractura de un aze??tropo con la manipulaci??n de presi??n unidireccional
Una destilaci??n al vac??o se puede utilizar para "romper" una mezcla azeotr??pica. La variaci??n de la temperatura del vapor generar matraz cuando destilaci??n un aze??tropo de fr??o hasta el punto de ebullici??n soluciones no producir una relaci??n de deslizamiento continua de producto a contaminar en el destilado. Siendo los dos puntos de ebullici??n separados siguen siendo, simplemente se superponen; estos pueden ser considerados como necesarios energ??as de activaci??n para la liberaci??n de un vapor particular. Al exponer a un aze??tropo a un vac??o, es posible sesgo el punto de ebullici??n de uno lejos del otro mediante la explotaci??n de la diferencia entre cada presi??n de vapor de los componentes. Cuando el sesgo es suficientemente grande, los dos puntos de ebullici??n ya no se superponen y por lo tanto la banda azeotr??pica desaparece.
Este m??todo no est?? exento de inconvenientes. Como ejemplo, la exposici??n de una soluci??n de agua y etanol a un vac??o de 70 torr permitir?? etanol absoluto a ser destilada. Sin embargo, debido a la atm??sfera de baja presi??n, el vapor de etanol requiere una superficie del condensador significativamente enfriador para licuar, al pasar de 78,3 ?? C a presi??n atmosf??rica a 24,5 ?? C a 70 torr; el no proporcionar dichos resultados en los vapores que pasan a trav??s del condensador y en la fuente de vac??o. Esto tambi??n puede afectar la eficiencia del condensador, como la temperatura de licuefacci??n de gotas hacia el m??nimo el equipo de condensaci??n puede enfriar a, el gradiente t??rmico a trav??s de las superficies de licuefacci??n y reduce, por lo que con ello, la velocidad a la cual el calor se puede extraer a partir del vapor .
Por el contrario, el aumento de una presi??n de destilaci??n tambi??n puede romper un aze??tropo, pero traer?? consigo la posibilidad de descomposici??n t??rmica, para compuestos org??nicos, en particular, y puede ser m??s beneficioso para destilaciones tolerantes a alta temperatura, tales como los de las sales met??licas.
La destilaci??n a presi??n oscilante
Este m??todo de destilaci??n se puede utilizar para separar mezclas azeotr??picas y se basa en un principio similar a destilaci??n a vac??o, que es la manipulaci??n de los puntos de ebullici??n mediante la alteraci??n de la presi??n de la atm??sfera a la que se expone una soluci??n.
Podr??a ser elegido m??s de destilaci??n al vac??o pura de un aze??tropo si esa soluci??n, por ejemplo, ten??a un punto de licuefacci??n tan baja a la presi??n requerida para romper el aze??tropo que el equipo fue incapaz de proporcionar para ello, permitiendo que el producto fluya hacia fuera de la condensador y en la fuente de vac??o. Aqu??, en lugar de manipular a s??lo un punto de ebullici??n, uno o m??s se ven alterados, uno tras otro; con el n??mero de alternancias de presi??n est?? determinada por el n??mero de componentes en la soluci??n de alimentaci??n considerado como contaminantes. Esto podr??a ser beneficioso para una purificaci??n, ya que es probable que crear necesidades t??rmicas menos extremas. Simplemente, en lugar de hacer pivotar presi??n de destilaci??n en una direcci??n sola en un intento de romper el aze??tropo en un solo paso, la ruptura se realiza en dos o m??s etapas con una presi??n moverse en dos para crear una banda de trabajo centrada en torno a las temperaturas m??s accesibles; quiz??s pasando de una presi??n negativa a la atmosf??rica y a una presi??n positiva. En esencia, la destilaci??n a presi??n oscilante es un intento de reducir las condiciones extremas mediante la dispersi??n de la carga a trav??s de la manipulaci??n del equipo que genera el entorno de destilaci??n.
Si se desea una alimentaci??n continua, o las presiones de destilaci??n requeridas son suficientemente extremo para justificar dise??o especializado, cada paso puede requerir una columna separada f??sicamente. Si s??lo se requiere una ejecuci??n de lote y la misma columna puede realizar bajo todas las presiones requeridas, esta sola columna puede ser suficiente; con el matraz de generaci??n de vapor se vac??a despu??s de la primera destilaci??n, la primera ejecuci??n destilado de nuevo al comienzo y la repetici??n de destilaci??n bajo las condiciones de presi??n segundo, y as?? sucesivamente.
La selecci??n de qu?? componente del destilado debe ser sesgada hacia puede hacerse bas??ndose en la energ??a requerida para evaporar desde la soluci??n de alimentaci??n.
Destilaci??n a presi??n oscilante se emplea durante la purificaci??n de acetato de etilo despu??s de su s??ntesis catal??tica de etanol.
Destilaci??n Industrial

Aplicaciones de destilaci??n industrial a gran escala incluyen tanto por lotes y fraccionada continua, vac??o, azeotr??pica, extractiva, y la destilaci??n de vapor. Las aplicaciones industriales m??s utilizados de continuo, destilaci??n fraccionada en estado estacionario est??n en las refiner??as de petr??leo , petroqu??mica y plantas qu??micas y plantas de procesamiento de gas natural.
Destilaci??n industrial se realiza normalmente en grandes columnas, cil??ndricos verticales conocidas como torres de destilaci??n o columnas de destilaci??n con di??metros que van de unos 65 cent??metros a 16 metros y alturas que van desde unos 6 metros a 90 metros o m??s. Cuando la alimentaci??n del proceso tiene una composici??n diversa, como en la destilaci??n de petr??leo crudo , salidas de l??quido a intervalos de hasta la columna permiten la retirada de las diferentes fracciones o productos que tienen diferentes puntos de ebullici??n o intervalos de ebullici??n. Los productos "ligeros" (aquellos con el punto de ebullici??n m??s bajo) de salida de la parte superior de las columnas y los productos "pesados" (aquellos con el punto de ebullici??n m??s alto) de salida de la parte inferior de la columna y se llaman a menudo los fondos.

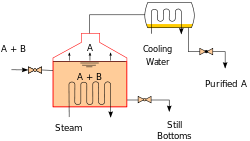
Torres industriales a gran escala utilizan reflujo para lograr una separaci??n m??s completa de productos. El reflujo se refiere a la parte del producto l??quido de cabeza condensada de una torre de destilaci??n o fraccionamiento que se devuelve a la parte superior de la torre como se muestra en el diagrama esquem??tico de un t??pico, a gran escala torre de destilaci??n industrial. Dentro de la torre, el l??quido de reflujo flujo descendente proporciona un enfriamiento y condensaci??n de los vapores que fluye hacia arriba, aumentando as?? la eficacia de la torre de destilaci??n. Se proporciona la m??s reflujo durante un n??mero dado de platos te??ricos, mejor es la separaci??n de la torre de los materiales de menor punto de ebullici??n a partir de materiales de punto de ebullici??n m??s altos. Alternativamente, se proporciona la m??s reflujo durante una separaci??n deseada dada, se requiere el menor n??mero de platos te??ricos.
Tales torres de fraccionamiento industrial tambi??n se utilizan en la separaci??n de aire, produciendo el l??quido de ox??geno , nitr??geno l??quido, y de alta pureza de arg??n . Destilaci??n de clorosilanos tambi??n permite la producci??n de alta pureza de silicio para su uso como un semiconductor .
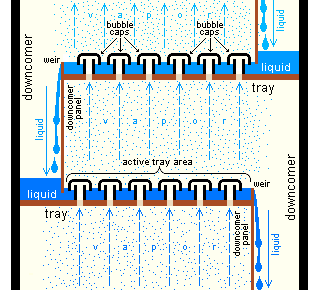
Dise??o y operaci??n de una torre de destilaci??n depende de la alimentaci??n y de los productos deseados. Dada una alimentaci??n simple componente, binario, m??todos anal??ticos como la M??todo de McCabe-Thiele o la Fenske ecuaci??n se puede utilizar. Para una alimentaci??n de m??ltiples componentes, modelos de simulaci??n se utilizan tanto para el dise??o y operaci??n. Adem??s, las eficiencias de los dispositivos de contacto de vapor-l??quido (referidos como "placas" o "bandejas") utilizados en torres de destilaci??n son generalmente m??s bajos que el de un te??rico 100% eficiente etapa de equilibrio. Por lo tanto, una torre de destilaci??n necesita m??s bandejas que el n??mero de etapas de equilibrio l??quido-vapor te??ricas.
En usos industriales, a veces un material de embalaje se utiliza en la columna en lugar de bandejas, especialmente cuando bajo presi??n cae a trav??s de la columna se requieren, como cuando se opera bajo vac??o.

Este material de envasado, se puede aleatoria arroj?? embalaje (1/3 "de ancho) como Anillos Raschig o l??mina de metal estructurada. Los l??quidos tienden a mojar la superficie de la empaquetadura y los vapores pasan a trav??s de esta superficie mojada, donde transferencia de masa se lleva a cabo. A diferencia de la destilaci??n de la bandeja convencional en el que cada bandeja representa un punto de equilibrio vapor-l??quido separado, la curva de equilibrio vapor-l??quido en una columna de relleno es continua. Sin embargo, cuando el modelado de columnas de relleno, es ??til para calcular un n??mero de "etapas te??ricas" para denotar la eficiencia de separaci??n de la columna de relleno con respecto a las bandejas m??s tradicionales. A diferencia envases con forma tienen diferentes ??reas de superficie y el espacio vac??o entre envases. Estos dos factores afectan el rendimiento de embalaje.
Otro factor en adici??n a la zona forma de embalaje y la superficie que afecta el rendimiento de relleno aleatorio o estructurado es la distribuci??n de l??quido y vapor que entra en el lecho empaquetado. El n??mero de etapas te??ricas requeridas para hacer una separaci??n dada se calcula utilizando un vapor espec??fico para relaci??n l??quido. Si el líquido y el vapor no se distribuyen de manera uniforme en toda la zona de la torre superficial, ya que entra en el lecho de relleno, el líquido a vapor relación no será correcta en el lecho de relleno y no se consigue la separación necesaria. El embalaje se parece no estar funcionando correctamente. La equivalente altura de un plato teórico (HETP) será mayor de lo esperado. El problema no es el embalaje en sí, sino la mala distribución de los fluidos que entran en el lecho relleno. Líquido mala distribución es más frecuente el problema de vapor. El diseño de los distribuidores de líquido utilizados para introducir la alimentación y el reflujo a un lecho de relleno es fundamental para hacer que el embalaje llevar a cabo para que la máxima eficiencia. Métodos de evaluar la eficacia de un distribuidor de líquido para distribuir uniformemente el líquido que entra en un lecho de relleno se pueden encontrar en las referencias. Un trabajo considerable que se ha hecho sobre este tema por el Fraccionamiento Research, Inc. (comúnmente conocido como FRI).
La destilación en la elaboración de alimentos
Bebidas destiladas
Carbohidratos materiales vegetales -containing se permite a fermentar, produciendo una solución diluida de etanol en el proceso. Espíritus como el whisky y el ron son preparados por destilación estas soluciones diluidas de etanol. Otros componentes que el etanol se recogen en el condensado, incluyendo el agua, ésteres, y otros alcoholes que representan el sabor de la bebida.