Cycle de l'azote
|
|
Cet article ne cite pas suffisamment ses sources (mars 2011). Si vous disposez d'ouvrages ou d'articles de référence ou si vous connaissez des sites web de qualité traitant du thème abordé ici, merci de compléter l'article en donnant les références utiles à sa vérifiabilité et en les liant à la section « Notes et références » (modifier l'article, comment ajouter mes sources ?).
|

Le cycle de l'azote est un cycle biogéochimique qui décrit la succession des modifications subies par les différentes formes de l'azote neutre en formes réactives (diazote, nitrate, nitrite, ammoniac, azote organique) et vice-versa.
Les analyses isotopiques de l'azote faites dans différents compartiments géologiques et de la biosphère et notamment dans les sédiments lacustres montrent que ce cycle a été récemment fortement perturbé par l'Homme qui a plus que doublé la quantité d'azote réactif (Nr) annuellement ajoutés à la biosphère, essentiellement à partir de 1895 ± 10 ans (± 1 pour l'écart-type) avec une forte augmentation dans les années 1960 à 2010, principalement dans l'hémisphère Nord[1].
Généralités
L'azote est un composé de base vital pour de nombreux processus biologiques dont la synthèse des acides aminés constituant les protéines et les bases nucléiques de l'ADN.
L'atmosphère contient une grande quantité de diazote (79 % en volume), mais cet azote gazeux est très peu biodisponible pour la plupart des espèces. Seules quelques bactéries et cyanobactéries (principalement présentes dans le sol), telles que Azotobacter vinelandii peuvent fixer l'azote atmosphérique. Elles le font grâce à un enzyme, la nitrogénase qui est capable de casser la triple liaison N≡N qui rend l'azote gazeux si stable, mais uniquement dans certains contextes, car cette réaction de transformation d'azote atmosphérique en une forme assimilable par les organismes a un coût énergétique et métabolique élevés, et fait appel à d'autres éléments nécessaires à la biosynthèse de la nitrogénase.
Pour ces raisons, l'azote biodisponible est un puissant facteur limitant pour la production de biomasse algale et végétale, et donc pour la productivité de nombreux écosystèmes. Ceci explique aussi que la disponibilité en azote joue un rôle majeur dans la structuration et les dynamiques d'évolution de nombreuses communautés végétales.
Les bactéries fixatrices de l'azote produisent de l'ammoniaque (NH4OH) à partir de l'azote atmosphérique et de l'hydrogène de l'eau (l'ammoniaque est le nom de la forme soluble dans l'eau du gaz ammoniac). Certaines de ces bactéries, comme Rhizobium, vivent en symbiose avec des plantes, produisant de l'ammoniaque. Dans les sols bien oxygénés, mais aussi en milieu aquatique oxygéné, des bactéries transforment l'ammoniac (NH3) en nitrite (NO2-), puis en nitrates (NO3-), au cours du processus de nitrification. On peut décomposer cette transformation en nitritation et nitratation. En contrepartie de ce service, les plantes symbiotes fournissent des glucides aux bactéries (dans la rhizosphère).
La décomposition d'organismes morts (nécromasse) par des bactéries saprophytes sous forme d'ions ammonium (NH4+) est une autre source d'ammoniaque.
Les végétaux absorbent (au travers des feuilles pour certaines plantes aquatiques et sinon via les racines les ions nitrate (NO3-) et, dans une moindre mesure, l'ammonium présent dans le sol. Ils seront principalement incorporés dans les acides aminés et les protéines. Les végétaux constituent ainsi la source primaire d'azote assimilable par les animaux.
En milieu anoxique (sol ou milieu aquatique non oxygéné), des bactéries dites dénitrifiantes transforment les nitrates en gaz diazote, c'est la dénitrification.
Le cycle naturel de l'azote a toutefois été fortement perturbé par les activités humaines, qui ont modifié la disponibilité de l'azote, avec des implications encore mal évaluées pour les écosystèmes à travers le monde.
L'azote géologique
Une partie, souvent oublié du cycle de l'azote passe par le substratum rocheux là où de l'azote était présent dans la matière organique sédimentée ou liée à cette dernière ; de l' « Azote géologique » est aussi trouvé dans certaines eaux thermales, provenant alors de roches sédimentaires, du manteau et parfois de sources météoriques[2].
Les taux d'azote dans les roches varient de simples traces (<200 mg N kg-1) dans les granites à des taux écologiquement important, dépassant 1000 mg N kg-1 dans certains et roches sédimentaires et métasédimentaires[3]. Des roches riches en azote sont présentes un peu partout dans le monde et pourraient aussi jouer un rôle local, sous-estimé, dans la disponibilité en azote pour le réseau trophique. En fait, selon Schlesinger (1997) près de 20% de la totalité du cycle de l'azote serait fixé dans les roches[4].
Ce rôle pourrait avoir été négligé en raison d'un manque de méthodes analytiques de routine pour la quantification de l'azote dans les roches. Les dépôts de nitrates accumulé dans les régions arides et semi-arides sont notamment des réservoirs potentiellement importants[2].
Dans la plupart des cas, les taux élevées d'azote dans l'eau et le sol ont une origine humaine. Parfois, elles peuvent être attribués aux effets d'intempéries sur le substrat rocheux (en cas de roche-mère riche en ammonium), avec dans certains environnements, une quantité d'azote libéré du socle rocheux supérieure aux besoins des plantes, et pouvant contribuer à saturer le milieu terrestre et l'écosystème local en produisant des phénomènes d'eutrophisation et de dystrophisation. L'oxydation du nitrate d'ammonium peut aussi contribuer à l'acidification des sols et au phénomène plus général d'acidification des eaux douces, susceptible d'inhiber la revégétalisation et renaturation spontanée de certains écosystèmes[2].
Il semble que les écosystème établis sur des roches à teneurs élevées en azote soient plus vulnérables à l'anthropisation ; plusieurs études récentes ont montré que l'azote géologique existe en quantité significative, qu'il peut être libéré et réactivé dans certaines circonstances, avec alors selon Holloway & Dahlgren (2002). « un potentiel d'amplification des impacts humains sur le cycle de l'azote dans les écosystèmes terrestres et aquatiques »[2].
L'azote géologique (outre les formes organiques de l'azote ou liée à la matière organique, surtout trouvées dans les sédiments organique jeunes), l'azote géologique et inorganique est notamment constitué des minéraux suivants :
| Minéral | Formule | Occurrence (exemples) |
|---|---|---|
| Osbornite | TiN | Chondrite[5] |
| Sinoite (en) | Si2N2O | Chondrites [6],[7] |
| Nitratine | NaNO3 | Désert d'Atacama (Chili) ; Désert de Mojave (États-Unis) ; Ouest des États-Unis ; Grottes (États-Unis)[8],[9],[10] |
| Nitre | KNO3 | Désert d'Atacama (Chili), Ouest des États-Unis ; Grottes (États-Unis)[8],[10]... |
| Nitrocalcite | Ca(NO3)2·4H2O | Dépôts dans des grottes, AZ, USA [11] |
| Svéite | KAl7(NO3)4Cl2(OH)16·8H2O | Dépôts dans des grottes (ex : Venezuela)[12] |
| Gerhardtite | Cu2(NO3)(OH)3 | Dépôts liés au minerai de cuivre (ex : Australie; Congo; AZ, USA)[13],[14],[15] |
| Likasite | Cu3(OH)5(NO3)·2H2O | Dépôts liés au minerai de cuivre (Congo)[16] |
| Darapskite | Na3(SO4)(NO3)·H2O | Origine hydrothermale (Antarctique)[17] ; Caliche : Désert d'Atacama (Chili) [8] ; Désert de Mojave (USA) [8] |
| Humberstonite | Na7K3Mg2(SO4)6(NO3)2·6H2O | Caliche : Désert d'Atacama (Chili), Désert de Mojave (USA)[18],[9] |
| Ammoniojarosite | (NH4)Fe3+(SO4)2(OH)6 | Minerais : WY & UT, USA; origines hydrothermales Geysers, CA[19],[20],[21] |
| Boussingaultite | (NH4)2Mg(SO4)2·6H2O | Origines hydrothermales Geysers (CA)[22],[23] |
| Lecontite | (NH4,K)Na(SO4)·2H2O | Évaporites (ex : Pakistan) [24] |
| Letovicite | (NH4)3H(SO4)2 | Origine hydrothermale : Geysers CA, USA ; Campi Flegrei (Italie)[21],[25] |
| Mascagnite | (NH4)2SO4 | Origine hydrothermale : Geysers, CA, USA[21] |
| Salmiac | (NH4)Cl | Source hydrothermale (ex : Etna et Vésuve en Italie)[26] |
| Tschermigite | (NH4)Al(SO4)2·12H2O | Source hydrothermale ; Geysers, CA. USA ; Campi Flegrei en Italie ; Taupo en Nouvelle-Zélande[21],[25],[27] |
| Mundrabillaite | (NH4)2Ca(HPO4)2·H2O | Dépôts dans des grottes (Australie)[28] |
| Ammonian fluorapophyllite | (NH4,K)Ca4Si8O20(F,OH)·8H2O | Source hydrothermale : Calvinia (Afrique du Sud) et Guanajuato (Mexique) [29] |
| Ammonioleucite | (NH4)AlSi2O6 | Roches métamorphiques volcaniques : Japon[30] ; Source hydrothermale : Geysers, CA[31] |
| Buddingtonite | NH4AlSi3O8 | Source hydrothermale : Sulphur Bank Mine, CA ; Cedar Mt, NV, USA ; Japon ; Phosphoria Fm.: ID, USA ; oil shale : Australia [32] ,[33],[34],[35],[36] |
| Tobélite | (NH4,K)Al2(Si3Al)O10(OH)2 | Argiles hydrothermiquement altérées : Japon; UT, USA ; pétrole de schiste en mer du nord[37],[38],[39] |
En mer
Sous forme de nitrate ou d'ammoniaque, l'azote est très soluble dans l'eau et mobile dans les écosystèmes. Le ruissellement, l'érosion et les pluies tendent à ramener les nitrates non captés par la biomasse terrestre vers les lacs et surtout vers les nappes phréatiques et les océans. Dans la mer, comme sur terre, l'azote dissous dans l'eau est capté (via la photosynthèse) par les plantes et certaines bactéries, puis concentré dans le réseau trophique sous forme de protéines animales notamment.
Tout poisson et tout animal marin concentre dans sa biomasse (matière organique) une partie de l'azote accumulé par les végétaux ou animaux qu’il a consommés, lesquels l’ont prélevé dans le stock océanique, alimenté par l’atmosphère, et de plus en plus excessivement par les apports terrigènes (engrais, et eaux usées essentiellement).
Depuis le développement de la pêche industrielle et intensive, cette quantité n’est plus négligeable. En effet, chaque poisson pêché en mer correspond donc à un retrait d’azote du compartiment océanique. Cette quantité a été évaluée pour 58 écosystèmes marins répartis sur le globe par une étude[40] récente sur le cycle de l'azote dans les régions côtières. En 1960 la pêche en mer ramenait vers la terre 60 % des composés azotés estimés être apportés par les fertilisants lessivés par les pluies. Or, de 1960 à 2004, ces apports azotés ont été multipliées par 7,5, alors qu’en raison d’une raréfaction des espèces commercialisables suite à la surexploitation des océans, les prises de pêche n’ont augmenté que de 2,5. Seulement environ 20 % de l’azote perdu en mer par l’agriculture sont donc en 2008 « récupérés » par les activités de pêche, laquelle devrait être pour ces raisons mieux prise en compte dans les calculs des bilans d’azote et risques d’eutrophisation.
Une interprétation facile et rapide de ces résultats serait de croire qu’en augmentant encore la pêche on diminuerait l’eutrophisation marine. Cette piste serait illusoire, rappelle Greenpeace l'auteur de l'étude elle-même, notamment parce que poursuivre la surpêche conduira rapidement à la disparition pure et simple de la plupart des espèces commerciales, et donc à une eutrophisation pire encore. La solution préconisée par les ONG environnementales et de nombreux organismes scientifiques, et notamment par un rapport récent[41] du Conseil de la recherche portant sur la « qualité de l'eau du fleuve Mississippi au regard de la Loi sur l'eau : progrès, défis et opportunités ». Ce rapport recommande que l'EPA et l'USDA coopérèrent mieux pour réduire les impacts de l'agriculture sur la qualité de l'eau du Mississippi et du nord du golfe du Mexique (zone morte de 22 000 km2 environ en 2007-2008 ; la plus vaste du monde). L'excès d'azote cause la prolifération de phytoplancton qui se décompose en consommant l'oxygène dissous dans l'eau aux dépens des organismes vivants qui disparaissent alors[42]. La seule solution durable serait de limiter la quantité de pertes d’engrais azotés d'origine agricole et de reconstituer un stock de poisson suffisant pour alimenter une pêche durable. Les indicateurs disponibles laissent penser que ces deux options peinent à se mettre en place.
Dans un aquarium
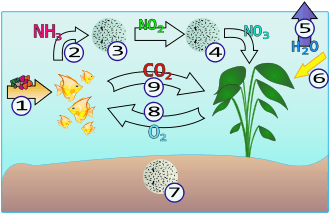
1 - Nourriture et nutriments
2 - Production d'urée et d'ammoniac
3 - Ammoniac → nitrites (Nitrosomonas)
4 - Nitrites → nitrates (Nitrospira)
5 - Évaporation
6 - Lumière
7 - Sol
8 - Oxygène
9 - Gaz carbonique
Dans un aquarium, ce sont les déjections des poissons et la nourriture non consommée qui sont à l'origine de la formation d'ammoniac. Ce produit extrêmement toxique pour les animaux aquatiques est transformé en nitrates beaucoup moins toxiques et bénéfiques pour les plantes par des bactéries présentes naturellement ou artificiellement (plantées) dans l'aquarium.
Les bactéries Nitrosomonas transforment l'ammoniac en nitrites, eux-mêmes très toxiques pour les animaux aquatiques. Et les bactéries Nitrobacter transforment les nitrites en nitrates. Nitrates qui sont alors absorbés par les plantes présentes dans l'aquarium.
Lors du premier remplissage de l'aquarium, ces bactéries sont absentes de l'eau. Les populations de bactéries apparaissent progressivement à partir du moment où l'aquarium est rempli. Le lancement du cycle nécessite, selon le contexte, entre trois et 8 semaines durant lesquelles l'eau de l'aquarium reste toxique. L'accumulation d'ammoniac provoque la prolifération de la première population de bactéries. Ces bactéries transforment l'ammoniac en nitrites. Au bout de quelques jours l'accumulation de nitrites due à la première population de bactéries provoque la prolifération de la deuxième population de bactéries, qui fait retomber la concentration de nitrites. La concentration de nitrites redescend jusqu'à être totalement indétectable. À partir de ce moment, l'aquarium est sain pour les poissons.
La plupart des aquariums ont un excédent de poissons, et pas assez de plantes par rapport à la quantité de nitrates produites. Les nitrates finissent par s'accumuler dans l'eau, et il est alors nécessaire de les diluer régulièrement au bout de une à deux semaines en remplaçant une part de l'eau de l'aquarium par de l'eau « neuve ». La fréquence des remplacements dépendra de la sensibilité des animaux. De nombreux poissons supportent des concentrations jusqu'à 50 mg/L. Par contre, les coraux « durs » ne supportent pas des concentrations supérieures à 2 mg/L.
Les bactéries dégradant l'ammoniac et les nitrites sont présentes dans l'eau, le sol de l'aquarium et le filtre. Le brassage provoqué par le filtre et la pompe amène l'eau riche en oxygène de la surface vers le fond, et assure la survie des bactéries - qui ont besoin d'oxygène pour vivre. Il sert également à mettre en contact les bactéries présentes dans le filtre avec l'eau de l'aquarium.
À partir du moment où le cycle de l'azote est en route, on dit que l'aquarium est cyclé : l'ammoniac et les nitrites qui se forment dans l'eau sont immédiatement transformés, et les concentrations restent à des niveaux indétectables.
Ammonification
C'est la production d'ammonium ou d'ammoniac du fait d'une activité biologique, à partir soit de matière organique en décomposition, soit par la réduction de nitrate.
À partir de la matière organique
L'ammonification à partir de la matière organique a pour but de produire une source d'azote directement utilisable par les organismes pour leur propre synthèse.
Réalisée uniquement par une biomasse microbienne :
- Bactéries gram- (Entérobactérie, Pseudomonacea, etc.)
- Bactéries gram+ (Bacillus, etc.)
- Bactéries gram+ (Entérocoques, etc.)
- Champignons
À partir des nitrates
Elle a pour buts :
- de produire de l'énergie par réduction des nitrates en ammonium.
- de produire une forme d'azote assimilable. C'est la dénitrification assimilatrice ou l'ammonification assimilatrice.
Application en traitement des eaux usées
Les stations d'épuration ont notamment comme fonction de dégrader les formes d'azote moléculaire et ammoniacal en nitrates, puis éventuellement de provoquer une dénitrification (ou dénitratation) afin de réduire la concentration de cet élément dans les eaux rejetées.
Nomenclature[43]
- HNO3 : acide nitrique
- HNO2 : acide nitreux
- NO-3 : ion nitrate
- NO2- : ion nitrite
- NH4+ : ion ammonium
- NH3 : ammoniac
- NH4OH : ammoniaque
- N2 : diazote
Illustrations

{kind=link}
Notes et références
- ↑ Gordon W. Holtgrieve & al (2011) “A Coherent Signature of Anthropogenic Nitrogen Deposition to Remote Watersheds of the Northern Hemisphere” Science Magazine 16 December 2011: 1545-1548 (résumé).
- 1 2 3 4 Holloway, J. M., & Dahlgren, R. A. (2002). Nitrogen in rock: occurrences and biogeochemical implications. Global Biogeochemical Cycles, 16(4), 65-1.
- ↑ Allègre, C., G. Manhés, and E. Lewin (2001) Chemical composition of the Earth and the volatility control on planetary genetics, Earth Planet. Sci. Lett., 185, 46–69.
- ↑ Schlesinger, W. H.. Biogeochemistry – An Analysis of Global Change, p. 588, Academic, San Diego, Calif., 1997.
- ↑ Story-Maskelyne, N., On the mineral constituents of meteorites, Philos. Trans. R. Soc. London, 160, 189–214, 1870.
- ↑ Mason, B., The enstatite chondrites, Geochim. Cosmochim. Acta, 30, 23–39, 1966.
- ↑ Keil, K., Mineralogical and chemical relationships among enstatite chondrites, J. Geophys. Res., 73, 6945–6976, 1968
- 1 2 3 4 Ericksen, G. E., (1981) Geology and origin of the Chilean nitrate deposits, U.S. Geol. Surv. Prof. Pap., 1188, 37,
- 1 2 Ericksen, G. E., J. W. Hosterman, and P. St. Amand, Chemistry, mineralogy, and origin of the clay-hill nitrate deposits, Amargosa River valley, Death Valley region, California, U.S.A. Chem. Geol., 67, 85–102, 1988.
- 1 2 Mansfield, G. R., and L. Boardman, Nitrate deposits of the United States, U.S. Geol. Surv. Bull., 838, 107, 1932.
- ↑ Hill, C. A., Mineralogy of Kartchner Caverns, Arizona, J. Cave Karst Stud., 61, 73–78, 1999
- ↑ Martini, J. E. J., Sveite, a new mineral from Autana Cave, Territorio Federal Amazonas, Venezuela, Trans. Geol. Soc. S. Afr., 83, 239–241, 1980.
- ↑ Wallace, M. E., and A. Pring, Gerhardtite, a copper hydroxy-nitrate from the Great Australia mine, Cloncurry, Queensland, Aust. Mineral., 5, 51–54, 1990.
- ↑ Toussaint, J., Sur la gerhardtite de Likasi, Bull. Soc. Geol. Belgique, 79, 233–235, 1956.
- ↑ Williams, S. A., Gerhardtite from the Daisy shaft, Mineral Hill mine, Pima County, Arizona, Ariz. Geol. Soc. Dig., 4, 123, 1961.
- ↑ Effenberger, E., Likasite, Cu3(OH)5(NO3)(2H2O: Revision of the chemical formula and redetermination of the crystal structure, Neues Jahrbuch Mineral. Monatsh., 1986, 101–110, 1986.
- ↑ Keys, J. R., Origin of crystalline, cold desert salts in the McMurdo region, Antarctica, Geochim. Cosmochim. Acta, 45, 2299–2306, 1981.
- ↑ Ericksen, G. E., J. J. Fahey, and M. E. Mrose, Humberstonite, Na7K3Mg2(SO4)(NO3)2(6H2O, a new saline mineral from the Atacama Desert, Chile, Geol. Soc. Am. Spec. Pap., 115, 59, 1968.
- ↑ Odum, J. K., P. L. Hauff, and R. A. Farrow, A new occurrence of ammoniojarosite in Buffalo, Wyoming, Can. Mineral., 20, 91–96, 1982
- ↑ Shannon, E. V., Ammoniojarosite, a new mineral of the jarosite group from Utah, Am. Mineral., 12, 424–426, 1927
- 1 2 3 4 Dunning, G. E., and J. F. Cooper Jr., History and minerals of The Geysers, Sonoma County, California, Mineral. Rec., 24, 339–354, 1993.
- ↑ Allen, E. T., and A. L. Day, Steam wells and other thermal activity at The Geysers, California, Carnegie Inst. Washington Publ., 378, 42, 1927.
- ↑ Goldsmith, E.. On boussingaultite and other minerals from Sonoma County, California, Acad. Nat. Sci. Philadelphia, 28, 264–266, 1877.
- ↑ Jan, M. Q., P. S. B. Colback, and M. Ahmad, Low-temperature secondary minerals from Tarbela, Geol. Bull. Univ. Peshawar, 18, 189–197, 1985.
- 1 2 Valentino, G. M., G. Cortecci, E. Franco, and D. Stanzione, Chemical and isotopic compositions of minerals and waters from the Camp Flegrei volcanic system, Naples, Italy, J. Volcanol. Geotherm. Res., 91, 329–344, 1999.
- ↑ Parwel, A., R. Ryhage, and F. E. Wickman, Natural variations in the relative abundances of the nitrogen isotopes, Geochim. Cosmochim. Acta, 11, 165–170, 1957.
- ↑ Martin, R., K. A. Rodgers, and P. R. L. Browne, The nature and significance of sulphate-rich, aluminous efflorescences from the Te Kopia geothermal field, Taupo Volcanic Zone, New Zealand, Mineral. Mag., 63, 413–419, 1999.
- ↑ Bridge, P. J., and R. M. Clark, Mundrabillaite - A new cave mineral from Western Australia, Mineral. Mag., 47, 80–81, 1983.
- ↑ Cavé, L. C., M. V. Fey, and D. K. Nordstrom, Dissolution rate of apophyllite: The effects of pH and implications for underground water storage, in Proceedings of the 10th International Symposium on Water Rock Interaction, Villasimius, Sardinia, Italy, edited by R. Cidu, pp. 251–254, A. A. Balkema, Brookfield, Vt., 2001.
- ↑ Hori, H. M., T. Nagashima, K. Yamada, and R. Miyawaki, Ammonioleucite, a new mineral from Tatarazawa, Fujioka, Japan, Am. Mineral., 71, 1022–1027, 1986
- ↑ Altaner, S. P., J. J. Fitzpatrick, M. D. Krohn, P. M. Bethke, D. O. Hayba, J. A. Goss, and Z. A. Brown, Ammonium in alunites, Am. Mineral., 73, 145–152, 1988
- ↑ Erd, R. C., D. E. White, J. J. Fahey, and D. E. Lee, Buddingtonite, an ammonium feldspar with zeolitic water, Am. Mineral., 49, 831–850, 1964.
- ↑ Krohn, M. D., Preliminary Description of a mineral-bound ammonium locality in the Cedar Mountains, Esmerelda County, Nevada, U.S. Geol. Surv. Open File Rep., 89-637. 12, 1989
- ↑ Kimbara, K., and T. Nishimura, Buddingtonite from the Toshichi Spa, Iwate Prefecture, Japan, J. Min. Soc. Jpn., 15, 207–216, 1982.
- ↑ Gulbrandsen, R. A., Buddingtonite, ammonium feldspar, in the Phosphoria Formation, southeastern Idaho, J. Res. U.S. Geol. Surv., 2, 693–697, 1974.
- ↑ Loughnan, F. C., F. I. Roberts, and A. W. Lindner, Buddingtonite, NH4 feldspar in the Condor Oilshale Deposit, Queensland, Australia, Mineral. Mag., 47, 327–334, 1983.
- ↑ Higashi, S., Tobelite, a new ammonium dioctahedral mica, Mineral J., 11, 16–27, 1982.
- ↑ Wilson, P. N., W. T. Parry, and W. P. Nash, Characterization of hydrothermal tobelitic veins from black shale, Oquirrh Mountains, Utah, Clays Clay Miner., 40, 405–420, 1992.
- ↑ Drits, V. A., H. Lindgreen, and A. L. Salyn, Determination of the content and distribution of fixed ammonium in illite-smectite by X-ray diffraction: Application to North Sea illite-smectite, Am. Mineral., 82, 79–87, 1997
- ↑ Étude conduite par la Pr. Roxane Maranger, du Département de sciences biologiques de l'Université de Montréal, publiée dans l’édition de février 2008 de Nature Geoscience)
- ↑ Mississippi River Water Quality and the Clean Water Act: Progress, Challenges, and Opportunities
- ↑ Henry Steinberg, greenpeace
- ↑ Toute la chimie 2ème période (2004) BALOU D., FABRITIUS E., GILLES A., Ellipses, Paris
Voir aussi
Articles connexes
- Réseau trophique
- Fixation biologique de l'azote
- Eutrophisation
- dystrophisation
- zone morte
- pédopaysage
- Acidification des océans
- Acidification des eaux douces
Liens externes
- Cycle de l'azote Schéma du cycle de l'azote en aquarium
- Cycle de l'Azote Explications sur la dégradation des composés azotés pour l'équilibre de l'aquarium.
Bibliographie
- James J. Elser (2011) A World Awash with Nitrogen Perspective Geochemistry Science 2011-12-16 Vol. 334 no. 6062 pp. 1504-1505DOI: 10.1126/science.1215567
 Portail de la biochimie
Portail de la biochimie  Portail de l’agriculture et l’agronomie
Portail de l’agriculture et l’agronomie  Portail de l’aquariophilie
Portail de l’aquariophilie