
{kind=link}

L'alignement multiple de s??quences
Contexte des ??coles Wikip??dia
Enfants SOS offrent un chargement complet de la s??lection pour les ??coles pour une utilisation sur les intranets des ??coles. parrainage SOS enfant est cool!
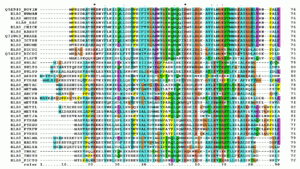
Un alignement de s??quences multiples (MSA) est un alignement de s??quences de trois ou plus des s??quences biologiques, g??n??ralement une prot??ine , ADN , ou ARN. En g??n??ral, l'ensemble des s??quences de requ??tes d'entr??e sont suppos??s avoir une ??volution relation par lequel ils partagent une lign??e et descendent d'un anc??tre commun. De l'MSA, s??quence r??sultante homologie peut ??tre d??duite et analyse phylog??n??tique peut ??tre men??e pour ??valuer origines ??volutives partag??es des s??quences. Repr??sentations visuelles de l'alignement comme dans l'image ?? droite illustrer ??v??nements de mutation telles que des mutations ponctuelles (d'un seul acide amin?? ou des modifications de nucleotides) qui apparaissent comme des caract??res diff??rents dans une colonne unique d'alignement, et l'insertion ou la deletion des mutations (ou indels) qui apparaissent comme des lacunes dans l'une ou plusieurs des s??quences dans l'alignement. Alignement de s??quences multiples est souvent utilis?? pour ??valuer s??quence conservation de des domaines de prot??ines, tertiaire et des structures secondaires, et les acides amin??s ou les nucleotides m??me individuels.
L'alignement multiple de s??quences se rapporte ??galement au proc??d?? d'alignement d'un tel ensemble de la s??quence. Parce que trois ou plusieurs s??quences de longueur biologiquement pertinente peut ??tre difficile et sont presque toujours du temps pour aligner ?? la main, de calcul des algorithmes sont utilis??s pour produire et analyser les alignements. MSA exigent des m??thodes plus sophistiqu??es que alignement par paires parce qu'ils sont plus de calculs complexes ?? produire. La plupart des programmes d'alignement de s??quences multiples utilisent plut??t que de m??thodes heuristiques optimisation globale, car l'identification de l'alignement optimal entre plus de quelques s??quences de longueur mod??r??e est prohibitivement co??teux en calcul.
La programmation dynamique et la complexit?? de calcul
La m??thode la plus directe pour produire un MSA utilise le technique de programmation dynamique pour identifier la solution globalement optimale d'alignement. Pour les prot??ines, ce proc??d?? implique g??n??ralement deux ensembles de param??tres: un p??nalit?? de gap et un matrice de substitution attribuant des notes ou des probabilit??s pour l'alignement de chaque paire possible des acides amin??s sur la base de la similitude des propri??t??s chimiques des acides amin??s et la probabilit?? d'??volution de la mutation. Pour des s??quences nucl??otidiques une matrice de substitution peut ??tre utilis??, mais comme il n'y a que quatre caract??res standard possibles par s??quence et les nucl??otides individuels ne diff??rent g??n??ralement pas beaucoup en remplacement probabilit??, les param??tres de s??quences d'ADN et d'ARN se composent g??n??ralement d'une p??nalit?? de br??che, positif score pour caract??re correspond, et un score n??gatif pour inad??quation.
Pour n s??quences individuelles, la m??thode n??cessite la construction de l'??quivalent de dimension n de la matrice form??e dans la programmation dynamique paires standard. L'espace de recherche augmente ainsi de fa??on exponentielle avec l'augmentation de n et est ??galement fortement d??pendant de la longueur de la s??quence. Pour trouver l'optimum global pour n s??quences de cette fa??on a ??t?? montr?? pour ??tre un Probl??me NP-complet. M??thodes pour r??duire l'espace de recherche en effectuant d'abord la programmation dynamique par paires sur chaque paire de s??quences dans le jeu de requ??te et la recherche que l'espace de solution ?? proximit?? de ces r??sultats (trouver efficacement l'intersection entre les chemins locaux entourant imm??diatement chaque solution optimale par paire) rendre la technique de programmation dynamique plus efficace. Le soi-disant "somme de paires" m??thode a ??t?? impl??ment??e dans le logiciel MSA, mais il est encore peu pratique pour de nombreuses applications qui n??cessitent la MSA alignement simultan?? de plusieurs dizaines, voire quelques centaines de s??quences. M??thodes de programmation dynamique sont maintenant utilis??s seulement quand un alignement tr??s haute qualit?? d'un petit nombre de s??quences est n??cessaire, et en tant que ??talonnage standard dans l'??valuation de nouvelles ou raffin??s techniques heuristiques.
La construction de l'alignement progressif
Un proc??d?? d'ex??cution d'une recherche heuristique d'alignement progressif est la technique (aussi connu comme le proc??d?? hi??rarchique ou arborescente) qui se accumule un MSA finale en effectuant d'abord une s??rie d'alignements par paires sur des s??quences successivement moins ??troitement apparent??es. Ces m??thodes commencent en alignant les deux s??quences les plus ??troitement li??es d'abord, puis en alignant successivement la s??quence suivante le plus ??troitement li?? ?? la requ??te mis ?? l'alignement produit dans l'??tape pr??c??dente. La paire "plus li??e" initial est d??termin?? par un syst??me efficace m??thode de classification tels que neighbor-joining bas??e sur une simple recherche heuristique de la requ??te sertie d'un outil comme FASTA. Techniques progressives donc construire automatiquement un arbre phylog??n??tique ainsi qu'un alignement.
Une limitation majeure des m??thodes progressives est leur forte d??pendance sur l'attribution initiale de parent?? et sur la qualit?? de l'alignement initial. Les m??thodes sont donc sensible aussi bien pour la distribution de s??quences dans l'ensemble de la requ??te; la performance se am??liore lorsque la parent?? entre les s??quences de requ??te est un gradient relativement lisse plut??t que lointainement grappes s??par??es. Les performances se d??gradent aussi consid??rablement lorsque toutes les s??quences dans le jeu sont plut??t apparent?? de loin, parce inexactitudes dans l'alignement initial sont alors plus susceptibles. La plupart des m??thodes progressistes modernes modifient leur fonction de score avec une fonction de pond??ration secondaire qui attribue des facteurs d'??chelle ?? des membres individuels de la requ??te d??finir de fa??on non lin??aire en fonction de leur distance phylog??n??tique de leurs voisins les plus proches. Choix judicieux de pond??ration peut aider ?? ??valuer la parent?? et att??nuer les effets de relativement pauvres alignements initiaux au d??but de la progression.
M??thodes d'alignement progressif sont suffisamment efficaces pour mettre en ??uvre sur une grande ??chelle pour de nombreuses s??quences et sont souvent ex??cut??s sur les serveurs Web publiquement accessibles afin que les utilisateurs ne ont pas besoin d'installer localement les applications d'int??r??t. Une m??thode tr??s populaire de l'alignement progressif est le Clustal famille, en particulier la variante pond??r??e ClustalW dont l'acc??s est fourni par un grand nombre de portails Web, y compris GenomeNet, EBI, et EMBnet. Diff??rents portails ou mises en oeuvre peuvent varier dans l'interface utilisateur et de faire diff??rents param??tres accessibles ?? l'utilisateur. Clustal est largement utilis?? pour la construction de l'arbre phylog??n??tique et comme entr??e pour la structure des prot??ines pr??vision par mod??lisation d'homologie.
Une autre m??thode de l'alignement progressif commun appel?? T-Coffee est plus lente que Clustal et ses d??riv??s, mais produit g??n??ralement alignements plus pr??cis pour jeux de s??quences apparent??es de loin. T-Coffee calcule alignements par paires en combinant l'alignement direct de la paire avec alignements indirects qui aligne chaque s??quence de la paire ?? une troisi??me s??quence. Il utilise la sortie de Clustal ainsi qu'un autre programme d'alignement local LALIGN qui distingue plusieurs r??gions de l'alignement local entre deux s??quences. L'alignement obtenu et arbre phylog??n??tique sont utilis??es comme guide pour la production de nouveaux et plus pr??cis des facteurs de pond??ration.
Comme les m??thodes progressistes sont heuristiques qui ne sont pas garantis ?? converger vers un optimum global, la qualit?? de l'alignement peut ??tre difficile ?? ??valuer et leur v??ritable signification biologique peut ??tre obscure. Une m??thode semi-progressive tr??s r??cente qui am??liore la qualit?? de l'alignement et ne pas utiliser une heuristique perte tout en fonctionnant en polynomiale a ??t?? mis en ??uvre dans le programme PSAlign.
M??thodes it??ratives
Un ensemble de m??thodes pour produire MSA tout en r??duisant les erreurs inh??rentes aux m??thodes progressistes sont class??s comme ??it??ratif?? parce qu'ils fonctionnent de mani??re similaire aux m??thodes progressistes mais r??aligner plusieurs reprises les s??quences initiales ainsi que l'ajout de nouvelles s??quences ?? la MSA croissante. L'une des raisons m??thodes progressives sont si fortement d??pendante d'un alignement initial de haute qualit?? est le fait que ces alignements sont toujours incorpor??s dans le r??sultat final - qui est, une fois qu'une s??quence a ??t?? align??e dans la MSA, l'alignement ne est pas examin??e plus avant. Cette approximation am??liore le rendement au d??triment de la pr??cision. En revanche, les m??thodes it??ratives peuvent revenir ?? alignements par paires pr??c??demment calcul??s ou sous-MSA incorporant des sous-ensembles de la s??quence de requ??te comme un moyen d'optimiser un g??n??ral fonction objectif comme trouver un score d'alignement de haute qualit??.
Une vari??t?? de subtilement diff??rentes m??thodes d'it??ration ont ??t?? mis en ??uvre et mis ?? disposition dans les logiciels; commentaires et des comparaisons ont ??t?? utiles mais g??n??ralement se abstenir de choisir une technique ??meilleur??. Le logiciel PRRN / PRRP utilise un algorithme hill-climbing d'optimiser son score d'alignement MSA et corrige de mani??re it??rative les deux poids d'alignement et les r??gions localement divergentes ou "Gappy?? de la MSA croissante. PRRP fonctionne mieux lorsqu'il affiner un alignement pr??c??demment construit par une m??thode plus rapide.
Un autre programme it??rative, dialign, adopte une approche inhabituelle de se concentrer ??troitement sur des alignements locaux entre les sous-segments ou s??quence motifs sans introduire une p??nalit?? de br??che. L'alignement des motifs individuels est alors obtenue avec une repr??sentation matricielle similaire ?? un terrain ?? matrice de points dans un alignement par paires. Un autre proc??d?? qui utilise des alignements locaux rapides que les points d'ancrage ou "germes" pour une proc??dure globale d'alignement plus lent est r??alis?? sous la Suite CHAOS / dialign.
Une troisi??me m??thode bas??e sur l'it??ration populaire appel?? MUSCLE (alignement de s??quences multiples par log-attente) am??liore sur les m??thodes progressives avec une mesure de distance plus pr??cis pour ??valuer le degr?? de parent?? des deux s??quences. La mesure de distance est mis ?? jour entre les ??tapes d'it??ration (bien que, dans sa forme originale, MUSCLE contenait seulement 2-3 it??rations selon que le raffinement a ??t?? activ??).
Mod??les de Markov cach??s
Mod??les de Markov cach??s sont des mod??les probabilistes qui peuvent affecter les probabilit??s de toutes les combinaisons possibles de lacunes, allumettes, et l'inad??quation de d??terminer la MSA le plus probable ou fix??s des CES possibles. HMM peuvent produire une seule sortie plus prolifique, mais peuvent aussi g??n??rer une famille de alignements possibles qui peuvent ensuite ??tre ??valu??s pour signification biologique. Parce que HMM sont probabiliste, ils ne produisent pas la m??me solution ?? chaque fois qu'ils sont ex??cut??s sur le m??me ensemble de donn??es; Ainsi, ils ne peuvent pas ??tre garantis ?? converger vers un alignement optimal. HMM peuvent produire des alignements ?? la fois globales et locales. Bien que les m??thodes ?? base de HMM ont ??t?? d??velopp??s relativement r??cemment, ils offrent des am??liorations significatives dans la vitesse de calcul, en particulier pour les s??quences qui contiennent des r??gions qui se chevauchent.
Typique HMM-m??thodes de travail en repr??sentant un MSA comme une forme de graphe acyclique orient?? connu comme un graphe partiel d'ordre, qui consiste en une s??rie de noeuds repr??sentant des entr??es possibles dans les colonnes d'un MSA. Dans cette repr??sentation une colonne qui est absolument conserv??es (ce est que toutes les s??quences dans le MSA part un caract??re particulier ?? une position particuli??re) est cod??e comme un noeud unique avec autant de connexions sortantes qu'il ya de caract??res possibles dans la colonne suivante de l'alignement. Dans les termes d'un mod??le de Markov cach?? typique, les ??tats observ??s sont les colonnes d'alignement individuels et les ??tats ??cach??s?? repr??sentent la s??quence ancestrale pr??sum??e ?? partir de laquelle les s??quences dans le jeu de requ??te sont ??mis l'hypoth??se d'avoir descendu. Une variante de recherche efficace du proc??d?? de programmation dynamique, connu sous le nom Algorithme de Viterbi, est g??n??ralement utilis?? pour aligner successivement le MSA croissante ?? l'autre dans la s??quence requ??te fix?? pour produire une nouvelle MSA. Cela est diff??rent de m??thodes d'alignement progressistes parce que le alignement de s??quences ant??rieures est mis ?? jour ?? chaque nouvel ajout de s??quence. Cependant, comme les m??thodes progressives, cette technique peut ??tre influenc??e par l'ordre dans lequel les s??quences dans le groupe de recherche sont int??gr??s dans l'alignement, en particulier lorsque les s??quences sont de parent?? ??loign??e.
Plusieurs logiciels sont disponibles dans lequel les variantes des m??thodes bas??es HMM-ont ??t?? mises en ??uvre et qui sont connus pour leur ??volutivit?? et l'efficacit??, bien que correctement en utilisant une m??thode de HMM est plus complexe que l'aide de m??thodes progressistes les plus courantes. Le plus simple est POA (Alignement partiel-Order); un proc??d?? similaire, mais plus g??n??ralis??e est mis en oeuvre dans l'emballage SAM (alignement de s??quences et de mod??lisation). SAM a ??t?? utilis?? comme source d'alignements pour la pr??diction de la structure des prot??ines ?? participer ?? la CASP exp??rience de pr??diction de la structure et de d??velopper une base de donn??es des prot??ines pr??dites dans les levures esp??ces S. cerevisiae. m??thodes de HMM peuvent ??galement ??tre utilis??s pour la recherche de base de donn??es avec HMMER.
Les algorithmes g??n??tiques et le recuit simul??
Les techniques standard d'optimisation en informatique - qui tous deux ont ??t?? inspir??s par, mais ne reproduisent pas directement, les processus physiques - ont ??galement ??t?? utilis??s dans le but de produire plus efficacement MSA qualit??. Une telle technique, algorithmes g??n??tiques, a ??t?? utilis?? pour la production MSA dans une tentative pour simuler largement le processus ??volutif hypoth??se qui a donn?? lieu ?? la divergence dans l'ensemble de la requ??te. Le proc??d?? fonctionne en brisant une s??rie de MSA en fragments possibles et en r??arrangeant les fragments de fa??on r??p??t??e ?? l'introduction de lacunes dans des positions variables. Un g??n??ral fonction objectif est optimis??e lors de la simulation, le plus g??n??ralement la ??somme de paires" fonction de la maximisation introduit dans les m??thodes de base MSA programmation dynamiques. Une technique pour les s??quences de prot??ines a ??t?? mis en ??uvre dans la saga de logiciel (alignement de s??quences par l'algorithme g??n??tique) et son ??quivalent dans l'ARN est appel?? RAGA.
La technique de recuit simul??, par lequel un produit MSA existant par un autre proc??d?? est affin??e par une s??rie de r??arrangements visant ?? trouver des r??gions plus optimale de l'espace d'alignement que celui de l'alignement d'entr??e occupe d??j??. Comme la m??thode des algorithmes g??n??tiques, recuit simul?? maximise une fonction objective comme la fonction de somme de paires. Recuit simul?? utilise un ??facteur de temp??rature" m??taphorique qui d??termine la vitesse ?? laquelle se d??roulent les r??arrangements et la probabilit?? de chaque r??arrangement; typiques des p??riodes alterne d'utilisation du taux de r??arrangement ??lev??s relativement faible probabilit?? (d'explorer les r??gions les plus ??loign??es de l'espace d'alignement) avec des p??riodes de baisse des taux et probabilit??s plus ??lev??es d'explorer plus ?? fond minima locaux pr??s des r??gions nouvellement "colonis??s". Cette approche a ??t?? mise en ??uvre dans le programme Msasa (Alignement multiple de s??quences par recuit simul??).
Motif conclusion

Motif constatation, ??galement connu sous le nom d'analyse de profil, est un proc??d?? de localisation s??quence motifs de MSA mondiaux qui est ?? la fois un moyen de produire une meilleure MSA et un moyen de produire une matrice de notation pour une utilisation dans la recherche d'autres s??quences pour des motifs similaires. Une vari??t?? de proc??d??s pour isoler les motifs ont ??t?? d??velopp??s, mais tous sont bas??s sur l'identification de motifs court hautement conserv??es au sein de l'alignement plus grande et la construction d'une matrice semblable ?? une matrice de substitution qui refl??te la composition d'acides amin??s ou de nucleotides de chaque position dans le motif putatif . L'alignement peut ensuite ??tre affin??e ?? l'aide de ces matrices. Dans l'analyse de profil standard, la matrice comporte des entr??es pour chaque caract??re possible, ainsi que des entr??es pour les lacunes. Alternativement, les algorithmes de recherche de mod??le statistiques peuvent identifier des motifs comme un signe pr??curseur d'une MSA plut??t que comme une d??rivation. Dans de nombreux cas lorsque l'ensemble de la requ??te contient seulement un petit nombre de s??quences ou de s??quences contient seulement tr??s li??s, pseudocounts sont ajout??s ?? normaliser la distribution refl??t??e dans la matrice de notation. En particulier, cette corrige les entr??es z??ro probabilit?? dans la matrice ?? des valeurs qui sont petites mais non nulle.
Blocs analyse est une m??thode de constatation motif qui limite motifs dans les r??gions sans br??ches dans l'alignement. Les blocs peuvent ??tre g??n??r??s ?? partir d'un MSA ou ils peuvent ??tre extraits ?? partir des s??quences non align??es au moyen d'un ensemble de motifs communs pr??calcul??e pr??c??demment g??n??r??e ?? partir de familles de g??nes connus. Bloquer le ballon se appuie g??n??ralement sur l'espacement des caract??res ?? haute fr??quence, plut??t que sur le calcul d'une matrice de substitution explicite. Le serveur BLOCS fournit une m??thode interactive pour localiser ces motifs dans les s??quences non align??es.
Statistique pattern-matching a ??t?? mis en ??uvre en utilisant ?? la fois le algorithme esp??rance-maximisation et la ??chantillonneur de Gibbs. Un des outils les plus courants de motifs d'enqu??te, appel??es MEME, utilise maximisation des attentes et des m??thodes de Markov cach??s pour g??n??rer des motifs qui sont ensuite utilis??s comme outils de recherche par son MAST de compagnon dans la suite combin??e MEME / MAST.