{kind=link}
Coagulation sanguine
|
|
Cet article ne cite pas suffisamment ses sources (avril 2013). Si vous disposez d'ouvrages ou d'articles de référence ou si vous connaissez des sites web de qualité traitant du thème abordé ici, merci de compléter l'article en donnant les références utiles à sa vérifiabilité et en les liant à la section « Notes et références » (modifier l'article, comment ajouter mes sources ?).
|
La coagulation sanguine est un processus complexe aboutissant à la formation de caillots sanguins. C’est une partie importante de l’hémostase où la paroi endommagée d’un vaisseau sanguin est couverte d’un caillot de fibrine pour arrêter l’hémorragie. Les troubles de la coagulation qui mènent à des risques de saignements plus importants sont appelés hémophilie. D'autres troubles de la coagulation peuvent mener à un plus grand risque de thrombose.
La coagulation est remarquablement préservée d'une espèce à l'autre : chez tous les mammifères, la coagulation repose sur la formation d'un clou plaquettaire et sur une composante protéique de coagulation (ce sont les facteurs de coagulation).
La coagulation débute presque instantanément après une brèche au niveau de la paroi endothéliale des vaisseaux sanguins. L'exposition du sang au facteur tissulaire initie des changements au niveau des plaquettes et du fibrinogène. Les plaquettes forment immédiatement un clou pour bloquer le saignement : c'est l'hémostase primaire. L'hémostase secondaire débute au même moment : des protéines plasmatiques appelées « facteurs de coagulation » réagissent dans une cascade complexe pour former des fibres de fibrine, qui renforcent le clou plaquettaire.
Le phénomène de coagulation sanguine est généralement modélisé mathématiquement par l'équation de Smoluchowski (en), qui permet notamment de modéliser la formation de caillots sanguins lors de l'occurrence du phénomène de gélation.
Physiologie
L'hémostase est l'ensemble des mécanismes permettant d'interrompre un saignement pour éviter l'hémorragie :
- la vasoconstriction diminue le calibre du vaisseau lésé ;
- l'hémostase primaire correspond à l’adhésion des plaquettes au vaisseau lésé et entre elles (agrégation plaquettaire) ;
- l'hémostase secondaire correspond à la coagulation proprement dite ;
- le caillot attire et stimule la croissance de fibroblastes et de cellules de muscle lisse au sein de la paroi vasculaire et entame le processus de réparation qui résultera finalement en la dissolution du caillot (fibrinolyse).
La formation du clou plaquettaire (ou hémostase primaire)
Lorsque l'endothélium est endommagé, le collagène de l'espace interstitiel, normalement isolé, est exposé aux plaquettes en circulation. Celles-ci se fixent directement au collagène avec des récepteurs glycoprotéiques Ia/IIa spécifiques au collagène. Cette adhésion est fortifiée par le facteur de von Willebrand (vWF), qui est relâché par les cellules endothéliales et les plaquettes. Le facteur de Von Willebrand forme des liens additionnels entre les fibres de collagène et les glycoprotéines Ib/IX/V des plaquettes. Ces adhésions activent les plaquettes.
Les plaquettes activées relâchent le contenu de leurs granules dans le plasma. Ces granules contiennent de l'ADP, de la sérotonine, le facteur d'activation plaquettaire, le facteur de Von Willebrand, le facteur plaquettaire 4 et le thromboxane A2, qui active des plaquettes additionnelles. Le contenu des granules active une cascade de récepteurs couplés aux protéines G, résultant en une concentration plus élevée de calcium dans le cytosol des plaquettes. Le calcium active la protéine kinase C, qui active à son tour la phospholipase A2. La phospholipase A2 modifie l'intégrine glycoprotéique IIb/IIIa, augmentant son affinité pour le fibrinogène. Les plaquettes activées changent de forme : de sphériques, elles deviennent stellaires, et le fibrinogène entrecroisant les glycoprotéines IIb/IIIa promeut l'aggrégation des plaquettes adjacentes (ce qui complète l'hémostase primaire).
La cascade de coagulation (ou hémostase secondaire)
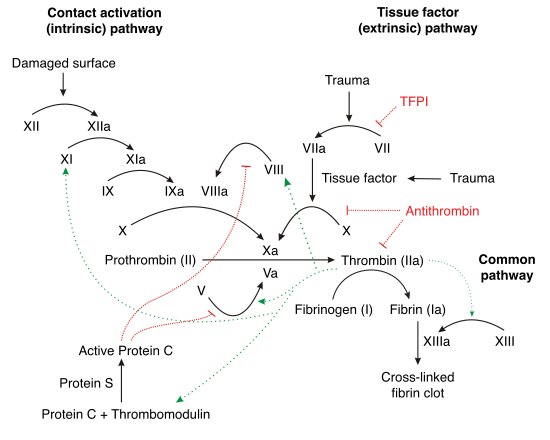
La cascade de coagulation est constituée de deux voies qui mènent à la formation de fibrine. Ces deux voies sont la voie extrinsèque (dépendante du facteur tissulaire) et la voie intrinsèque. On croyait auparavant que ces deux voies étaient d'importance égale dans la cascade de coagulation. On sait à présent que la voie la plus importante dans l'initiation de la coagulation est la voie extrinsèque. Les deux voies sont des séries de réactions dans lesquelles un zymogène de sérine protéase et son cofacteur glycoprotéique sont activés pour ensuite catalyser la prochaine réaction. Les facteurs de coagulation sont normalement identifiés par des chiffres romains, avec un a minuscule pour indiquer la forme active.
Les facteurs de coagulation sont généralement des sérine protéases (enzymes). Il y a quelques exceptions. Par exemple, le facteur VIII et le facteur V sont des glycoprotéines, et le facteur XIII est une transglutaminase. Les sérine protéases fonctionnent en clivant d'autres protéines à des résidus spécifiques de sérine. Les facteurs de coagulation circulent sous formes de zymogènes inactifs. La cascade de coagulation est classiquement divisée en trois voies : la voie extrinsèque et la voie intrinsèque activent tous les deux la voie commune finale du facteur X, de la thrombine et de la fibrine.
Voie extrinsèque
Le rôle principal de la voie extrinsèque est de générer très rapidement une grande quantité de thrombine. Le facteur VIIa circule dans des quantités plus importantes que tout autre facteur de coagulation activé.
- L'intégrité de la paroi des vaisseaux sanguins compromise, le facteur VII quitte la circulation et entre en contact avec le facteur tissulaire exprimé par les fibroblastes du stroma et par les leucocytes. Il y a formation du complexe activé TF-FVIIa.
- Le TF-FVIIa active le facteur IX et le facteur X.
- Le facteur VII est lui-même activé par la thrombine, par le facteur XIa, XII et Xa.
- L'activation du facteur X par le complexe TF-FVIIa est inhibée par l'inhibiteur de la voie du facteur tissulaire (TFPI).
- Le facteur Xa et son cofacteur Va forment le complexe prothrombinase, qui active la prothrombine en thrombine.
- La thrombine active alors d'autres facteurs de la cascade, dont le facteur V le facteur VIII et le facteur XI.
- Le facteur VIIIa est le cofacteur du facteur IXa, et leur complexe (appelé tenase) active le facteur X, puis le cycle recommence.
Voie intrinsèque
L'initiation de cette voie se fait par contact du sang avec les structures sous-endothéliales. Cette étape, appelée phase contact, fait intervenir le kininogène de haut poids moléculaire (KHPM), la prékallicréine et le facteur XII. Ce dernier activé, va lui-même activer le facteur XI en présence d'ions Ca++. En présence du XIa, le facteur IX est à son tour activé en IXa. Il se forme alors un premier complexe à la surface de la membrane plaquettaire capable d'activer le X en Xa. Ce complexe fait intervenir le IXa, le Ca++, le facteur 3 plaquettaire et le cofacteur VIII activé par les premières traces de thrombine.
Voie commune
La facteur Xa adsorbé à la surface des phospholipides d'origine tissulaire et plaquettaire va, en présence du facteur Va, constituer la prothrombinase. Le Va provient du V activé par la thrombine. La prothrombinase est donc un complexe enzymatique faisant intervenir le Xa, le Va, le Ca++ et des phospholipides. Il existe donc une similitude avec le complexe activateur du X. La prothrombinase permet la formation de thrombine (IIa) à partir de prothrombine (II).
Régulation de la coagulation
Différents mécanismes interviennent pour freiner et pour inverser la coagulation lorsque le caillot n'est plus nécessaire :
Inhibiteurs de la coagulation
| Inhibiteurs de la coagulation | |
|---|---|
| Nom | Fonction |
| Antithrombine | inhibe IIa, Xa |
| Protéine C | inactive Va et VIIIa |
| Protéine S | cofacteur de la protéine C |
| Tissue factor pathway inhibitor (TFPI) | inhibe le complexe facteur tissulaire - facteur VIIa et Xa. |
| Inhibiteur dépendant de la protéine Z (ZPI) | inhibe X et XI. |
| Protéine Z | cofacteur de ZPI |
| Heparin cofactor II | inhibe IIa |
Voir la revue[1] d'inhibiteurs physiologiques de la coagulation, d'autres anticoagulants dont oraux (anti- Vitamine K, ...; usages...).
Fibrinolyse
La fibrinolyse est le processus par lequel la fibrine est dégradée par la plasmine et est dissoute. La plasmine est activée au départ du plasminogène par l'urokinase et le tissue plasminogen activator.
Des troubles à ce niveau engendrent une hypercoagulabilité responsable d'un risque de thrombose.
Exploration de la coagulation par les tests de laboratoire
Exploration de l’hémostase primaire
- Temps de saignement
- Numération des plaquettes
- Dosage du facteur de von Willebrand
Exploration de l’hémostase secondaire
- Temps de céphaline activé (APTT, Temps de céphaline kaolin)
- Taux de prothrombine (TP, Temps de Quick, INR)
- Temps de thrombine - Temps de reptilase
- Dosage des facteurs de coagulation
Troubles de l’hémostase
Pathologie de l’hémostase primaire
- Thrombocytopénie
- Thrombopathie
- Maladie de von Willebrand
Pathologie de l’hémostase secondaire
- Hémophilies
- Déficit en vitamine K
- Insuffisance hépato-cellulaire
- les anticorps anti facteurs
Pathologie des inhibiteurs de la coagulation
Facteurs de coagulation
| Facteurs de coagulation et substances apparentées | ||
|---|---|---|
| N° | Nom | Fonction |
| I | Fibrinogène | forme des caillots (fibrine) |
| II | Prothrombine | active I, V, VIII, XI, XIII, protéine C, plaquettes. Vitamine K dépendant |
| III | Thromboplastine | cofacteur VIIa |
| IV | Calcium | |
| V | Proaccélérine | cofacteur X. |
| VI | (accélérine, ancien nom du Facteur Va) | |
| VII | Proconvertine | active IX, X. Vitamine K dépendant |
| VIII | Facteur anti-hémophile A | cofacteur IX |
| IX | Facteur Christmas ou facteur anti-hémophile B | active X. Vitamine K dépendant |
| X | Facteur Stuart-Prower | active II. Vitamine K dépendant |
| XI | Facteur Rosenthal, Antécédent de la thromboplastine plasmatique | active XII, IX et prékallikréine |
| XII | Facteur Hageman | active prékallikréine et fibrinolyse |
| XIII | Facteur fibrin-stabilizing | crosslinks fibrin |
| Facteur de von Willebrand | lie VIII, intermédiaire de l’adhésion des plaquettes | |
| prékallikréine ou Facteur Fletcher | active XII et prékallikréine ; scinde HMWK | |
| Kininogène de haut poids moléculaire (HPMK) | soutient l’activation réciproque de XII, XI, et prékallikréine | |
| fibronectine | médiateur adhésion cellulaire | |
Notes et références
Voir aussi
Article connexe
Liens externes
 Portail de la médecine
Portail de la médecine  Portail de l'hématologie
Portail de l'hématologie