{kind=link}
Pharmacocinétique
|
|
Cet article est une ébauche concernant la pharmacie, la biochimie et la médecine. Vous pouvez partager vos connaissances en l’améliorant (comment ?) selon les recommandations des projets correspondants.
|
La pharmacocinétique, parfois désignée sous le nom de « l'ADME » (voir plus loin) et qui suit la phase biopharmaceutique, a pour but d'étudier le devenir d'une substance active contenue dans un médicament après son administration dans l'organisme. Elle comprend, après la phase biopharmaceutique précédant le premier passage trans-membranaire, quatre grandes étapes:
- absorption (A) ;
- distribution (D);
- métabolisme (M);
- excrétion du principe actif et de ses métabolites (E).
La pharmacocinétique peut aussi concerner le devenir de substances chimiques non endogènes quelconques (ou xénobiotiques) dans le corps. Lorsque ces substances sont toxiques, ou données à des doses toxiques, on utilise alors le terme de « toxicocinétique ». Les études de toxicocinétique désignent aussi, en développement pharmaceutique non-clinique, les études de pharmacocinétique après administration répétée chez l'animal, au cours desquelles on étudie l’apparition d'effets secondaires au cours du temps.
La pharmacocinétique est comme la pharmacodynamie une sous-discipline de la pharmacologie. Un aphorisme la définit comme « ce que fait l'organisme à un médicament', tandis que la pharmacodynamique est définie comme 'ce que le médicament fait à l'organisme. »
La détermination des paramètres pharmacocinétiques d'une substance active apporte les informations qui permettent de choisir les voies d'administration et la forme galénique, et de déterminer le schéma posologique optimal (dose, fréquence d'administration) pour son utilisation future.
Pharmacocinétique descriptive : les processus de l'ADME
L'acronyme international ADME, qui est souvent utilisé pour désigner la pharmacocinétique, renvoie aux quatre grands processus qui la composent : Absorption, Distribution, Métabolisme, et Elimination. Un autre sigle international couramment utilisé pour désigner la pharmacocinétique est "PK" pour l'anglais : pharmacokinetics.
L'acronyme ADME peut entraîner une erreur d'interprétation dans un déroulement supposé strictement séquentiel des étapes de la pharmacocinétique d'un médicament.Si une molécule prise individuellement va passer successivement à travers les diverses étapes de l'ADME dans un ordre séquentiel, lorsque l'on s'intéresse à la globalité d'une prise médicamenteuse les étapes A, D, M et E se produisent de façon concomitante et dynamique. Une partie de la dose d'un médicament administré par voie orale peut ainsi être déjà filtrée au niveau glomérulaire (Elimination) alors que la totalité de la dose n'a pas encore été entièrement résorbée au niveau digestif (Absorption). À un instant t, les phases A, D, M et E se déroulent donc de façon simultanée au sein de l'organisme.
Libération (phase biopharmaceutique)
Le principe actif doit être libéré de sa forme galénique ou pharmaceutique afin de se rendre jusqu'au site de résorption. La forme galénique (ex. : capsule, solution, gélule, forme à libération prolongée, etc.) conditionne :
- la vitesse de dissolution et la mise en solution, ce qui affectera la vitesse de résorption du principe actif ;
Il s'agit d'une étape appelée également phase biopharmaceutique, et qui est critique essentiellement lorsqu'on considère les formes orales. La phase biopharmaceutique dépend essentiellement des choix réalisés en pharmacotechnie (choix du support galénique, type de matrices, enrobages et excipients utilisés). Cette étape ne peut être considérée comme une phase de pharmacocinétique stricto sensu car la molécule n'a pas encore passé sa première barrière biologique, quand bien même elle a été administrée per os : la lumière digestive est en effet considérée comme extérieure à l'organisme.
Absorption
Une fois libérée de son support galénique, la substance active doit traverser les membranes biologiques du site d'absorption vers le sang pour pénétrer dans la circulation systémique. L'absorption est la résultante de deux phénomènes: la résorption (passage membranaire) et les effets de premier passage (biotransformation métabolique survenant entre l'administration et la distribution générale).
La résorption des molécules thérapeutiques se fait le plus souvent par diffusion passive, dont la vitesse est gouvernée par la loi de Fick. Cette loi indique que la vitesse est inversement proportionnelle à l'épaisseur de la membrane considérée, et dépend de la liposolubilité du composé (Ks), de la surface d'échange, ainsi que du différentiel de concentration de part et d'autre de la membrane.
Cette loi permet donc de déduire intuitivement que l'intestin grêle (grande surface, faible épaisseur, forte vascularisation assurant un drainage important de l'autre coté de la membrane) présente les caractéristiques assurant une résorption maximale après administration orale d'un médicament).
Quatre points sont à considérer en particulier :
- les obstacles à franchir pour passer dans la circulation systémique : il s'agit des membranes biologiques, de type lipidique, comme :
- la paroi du tube digestif lors de l'administration de formulations orales : comprimés, solutions ou suspensions buvables, etc.
- la peau lors de l'administration de formulations percutanées : pommades, patchs, etc.
- la paroi du rectum lors de l'administration de suppositoires,
- la paroi du vagin lors de l'administration de formulations vaginales,
- la paroi des poumons pour les formes inhalées,
- etc.
- les conditions d'absorption relatives au médicament absorbé :
- la substance active doit être suffisamment hydrosoluble pour être dissoute,
- la substance active doit être suffisamment liposoluble pour franchir les barrières biologiques lipophiles,
- la substance active doit être sous forme neutre pour franchir les barrières biologiques par voie passive,
- la taille des molécules du principe actif peut aussi avoir son importance,
- la prise en charge ou non par des protéines de transport relevant d'une diffusion facilitée (SLC: protéines d'influx OATP, PEPT)ou d'un transport actif (famille ABC comme la p-glycoprotéine P ou les diverses isoformes de MRP, participant à l'efflux des molécules)
- etc.
- conditions d'absorption relatives au milieu :
l'équation de Henderson-Hasselbalch gouverne l'état d'ionisation d'une molécule présentant un pKa donné, en fonction du pH du milieu dans lequel elle se trouve. Cette loi permet de déduire le degré d'ionisation de la molécule, et donc sa capacité ou non à diffuser à travers une membrane biologique par voie passive ;- absorption dans l'estomac : les substances actives acides faibles sont absorbées au niveau de l'estomac, car elles ne sont pas ionisées en milieu acide ; sous forme non-ionisée liposoluble, elles sont par conséquent capables de franchir les barrières biologiques lipidiques. Les substances actives bases faibles sont elles faiblement absorbées au niveau de l'estomac, car elles sont ionisées en milieu acide,
- absorption au niveau de l'intestin grêle (milieu dont le pH varie de 5 à 8). Au vu du pKa de la majorité des molécules pharmacologiquement actives, le range de pH que l'on retrouve dans l'intestin grêle fait qu'un grande majorité de médicaments bases faibles ou acides faibles se retrouveront sous forme majoritaire non-ionisée et pourront ainsi être résorbés par diffusion passive.
- l'importance de la vidange gastrique. Si elle est trop rapide, les médicaments absorbés par l'estomac n'auront pas suffisamment de temps pour franchir les parois de l'estomac ; si elle est trop lente, les médicaments absorbés par l'intestin grêle parviendront dans le milieu sanguin avec un certain retard,
- la notion de premier passage :
Lors de la prise orale d'un médicament, suivie d'une absorption gastro-intestinale, la substance active est d'abord transportée par la veine porte vers le foie avant d'atteindre la circulation générale, et donc avant d'atteindre les sites d'actions au niveau des organes. La substance active peut ainsi subir une métabolisation hépatique pré-systémique avant d'atteindre la circulation générale : c'est l'effet de premier passage hépatique. C'est en général le plus important quantitativement, mais il existe également un premier passage digestif (action de l'équipement enzymatique des entero-bactéries et parfois des enzymes des entérocytes) et un premier passage pulmonaire. L'importance de chacun est différente selon les caractéristiques physico-chimiques et biologiques de la substance en question.
L'organisation du système vasculaire permet d'éviter en grande partie l'effet de premier passage hépatique en cas d'administration par suppositoire (voie rectale) : un tiers seulement du sang circulant dans les veines rectales passe par le foie avant d'atteindre la circulation générale. Ce premier passage est totalement évité par administration sub-linguale (médicament à faire fondre sous la langue) : la substance active est absorbée et déversée dans les veines jugulaires, aboutissant directement dans la circulation au niveau de l'arc aortique. Cette caractéristique fait une grande partie de l'intérêt de ces modes d'administration en cas d'urgence relative (un quart d'heure à une demi-heure de délai d'action). La voie inhalée et la voie transdermique évitent aussi en partie l'effet de premier passage hépatique.
Enfin, l'administration d'un principe actif par injection intraveineuse permet un passage direct dans la circulation, sans avoir à se soucier des problèmes liés à l'absorption évoqués ci-dessus. Ceci en fait la voie d'administration privilégiée dans les cas d'urgence. Les formes sublinguales permettent également de shunter la problématique liée à la mauvaise résorption et au premier passage hépatique de certaines molécules.
La phase d'absorption se caractérise par un paramètre pharmacocinétique appelé Biodisponibilité du principe actif, qui est le pourcentage de molécule administrée atteignant effectivement la circulation générale. Ce paramètre rend compte des phénomènes de résorption trans-membranaires et de premiers passages digestif ou et/ou hépatique. La biodisponibilité d'une voie IV est de 100 %, la biodisponibilité des formes orales peut varier de 5 % à 100 % en fonction des paramètres de résorption digestive et du poids du premier passage hépatique ou digestif.
Distribution
La substance active parvenue à la circulation systémique peut se lier plus ou moins fortement et de façon réversible aux protéines plasmatiques (albumine, alpha-1 glycoprotéine, lipoprotéines) :
- exemples de médicaments fortement liés aux protéines plasmatiques : dicoumarol (anticoagulant), phénytoïne (anti-épiléptique) ;
- exemples de médicaments moyennement liés aux protéines plasmatiques : phénobarbital (antiépileptique, hypnotique), théophylline (anti-asthmatique)… ;
- exemples de médicaments faiblement liés aux protéines plasmatiques : digoxine (cardiotonique), paracétamol (analgésique, antipyrétique)…
La molécule se retrouve dans le flux sanguin sous forme libre ou sous forme liée. Seule la forme libre pourra passer les membranes biologiques et exercer ultérieurement un effet pharmacodynamique. La forme liée est une forme de stockage et de transport, qui va se dissocier pour libérer la molécule sous forme libre selon la loi d'action de masse.
Certains médicaments peuvent également se lier aux éléments figurés du sang (érythrocytes) où ils sont séquestrés.
En fonction de ses propriétés physico-chimiques et biochimiques, la substance active peut également s'accumuler dans certains organes ou tissus contenant ou non des récepteurs pharmacologiques :
- affinité pour les protéines tissulaires ;
- affinité pour les acides nucléiques ;
- affinité pour les graisses ;
- affinité pour les tissus cibles contenant les récepteurs pharmacologiques.
On peut notamment calculer le volume de distribution de la substance pour caractériser quantitativement la distribution dans l'organisme d'un médicament. Au plus le volume est important, au plus la molécule aura tendance à quitter le flux vasculaire pour diffuser dans l'organisme, augmentant la probabilité que le tissu siège de la pathologie soit exposé à la substance active. Certains organes tel le cerveau sont toutefois protégés de l'effet de xénobiotiques potentiellement toxiques par des structures spécifiques telle la barrière hémato-encéphalique. Ces structures peuvent rendre problématique l'accès des substances actives aux récepteurs pharmacodynamiques à atteindre. L'essor de la nanomédecine (application des nanotechnologies aux sciences pharmaceutiques) vise essentiellement à améliorer la Distribution des substances actives en la rendant plus spécifique envers les tissus associés à la pathologie à traiter.
Métabolisation
C’est la transformation du médicament par le système enzymatique de l’organisme, en particulier au niveau du foie (organe contenant un équipement enzymatique riche et varié). Généralement, la biotransformation aboutit à des métabolites plus hydrophiles via des réactions de fonctionnalisation. Le gain en hydrophilie permet à l'organisme d'éliminer plus facilement par voie urinaire le médicament, sous forme de métabolites qui seront filtrés au niveau glomérulaire.
Les métabolites ainsi produits peuvent alors avoir :
- aucune activité (directement éliminés) ;
- une activité pharmacologique avant élimination ;
- une activité toxique avant élimination.
Les substances métabolisées peuvent être transformées en un autre produit actif. Ce phénomène peut être exploité pour concevoir un médicament inactif sous sa forme administrée au patient, mais conduisant à des métabolites actifs. On parle alors de médicament pro-drogue. Cet artifice peut être particulièrement utile quand la forme active de la substance active présente des difficultés d'administration ou d'absorption telle quelle. La capécitabine (Xeloda) est un exemple de pro-drogue orale utilisée en oncologie digestive et mammaire.
Élimination
Lorsqu'elle est présente dans la circulation générale, la substance active du médicament a tendance à être évacuée par l'organisme, qui utilise de nombreux mécanismes d'élimination (excrétion et/ou métabolisation). La substance active est éliminée par l'organisme soit sous forme inchangée, soit sous forme d'un ou de plusieurs métabolites généralement inactifs, soit encore sous les deux formes, dans des proportions variables. Cette “auto-épuration” de l'organisme se quantifie par le paramètre de clairance.
Clairance (CL)
Tout organe présente un courant d'entrée (flux artériel) et un courant de sortie (flux veineux). La présence d'une concentration médicamenteuse moindre au niveau veineux qu'au niveau artériel prouve que l'organe a éliminé une partie du médicament. Cette capacité d'épuration de l'organe correspond au processus de la clairance. On définit généralement la clairance sanguine ou plasmatique d'un médicament par un organe comme le volume sanguin ou plasmatique totalement débarrassé de la substance par unité de temps. Ce paramètre correspond à un débit exprimé en ml/min
La clairance est caractérisée par :
- la concentration du médicament à l'entrée de l'organe CA (sang artériel) ;
- la concentration du médicament à la sortie de l'organe CV (sang veineux) ;
- le débit sanguin Q de l'organe concerné.
Dès lors, la réaction médicament/organe s'exprime en termes de clairance selon la relation suivante :
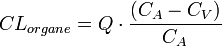
Clairance totale
L'épuration d'un médicament par l'organisme n'est généralement pas le fait d'un seul et unique organe. En effet, les possibilités pour l'organisme de se débarrasser d'une substance médicamenteuse sont multiples.
Deux mécanismes peuvent néanmoins être envisagés :
- l'élimination directe du composé ;
- l'élimination du composé après biotransformation (métabolisation).
La métabolisation d'une substance médicamenteuse est en règle générale principalement due au foie. Cependant, de nombreux autres organes peuvent y jouer un rôle : poumons, intestin ou cerveau.
Les voies d'excrétion sont elles aussi multiples. De manière logique, les voies prédominantes sont généralement la voie urinaire ou la voie biliaire, le plus souvent sous forme de métabolites conjugués (fécès). Cependant, de nombreux autres organes peuvent participer à cette élimination : les poumons, la peau (transpiration), les glandes salivaires, les glandes lacrymales…
Ainsi, un médicament subit généralement plusieurs processus d'épuration. La clairance totale correspond à la somme des clairances partielles (rénale, hépatique, intestinale, pulmonaire…). Très schématiquement, on distingue la clairance rénale et la clairance extrarénale.
La clairance extrarénale est le résultat :
- de la clairance hépatique provenant de l'addition des valeurs de clairance biliaire et de la clairance métabolique hépatique ;
- des différentes clairances métaboliques liées à l'activité de biotransformation des poumons, des intestins ou d'autres organes.
En résumé, pour l'élimination
Le foie est le site principal de la biotransformation de la substance active : métabolisation de la substance active en dérivés hydrosolubles, actifs ou inactifs. Dans une moindre part, il constitue un organe d'excrétion (élimination biliaire).
Le rein est le principal organe d'excrétion de la substance active inchangée et des métabolites plus hydrosolubles :
- filtration glomérulaire de la fraction libre du médicament ou de ses métabolites ;
- sécrétion tubulaire ;
- réabsorption.
Pharmacocinétique analytique : concepts et modèles
Influence de la dose administrée
L'effet pharmacologique ou thérapeutique d'un médicament peut être relié à l'évolution des concentrations plasmatiques au cours du temps. Cette relation dite PK/PD (pharmacocinétique-pharmacodynamique) permet de définir :
- un seuil thérapeutique : concentration minimale en dessous de laquelle aucune activité n'est obtenue ;
- une limite supérieure : concentration maximale au-delà de laquelle apparaissent des effets indésirables ;
- un intervalle thérapeutique : zone intermédiaire dans laquelle les concentrations sont à la fois actives et non-toxiques.
L'importance de l'écart entre les concentrations actives et toxique est variable. Cet écart est appelé index thérapeutique. Ainsi, on distingue les médicaments à index thérapeutiques élevé et les médicaments à index thérapeutique faible. Pour ces derniers, les posologies d'administration doivent être bien définies afin que les concentration plasmatiques restent strictement dans l'intervalle thérapeutique. C'est le cas du lithium, de la phénytoïne, de la digoxine, de la théophylline, etc.
La pharmacocinétique d'une substance médicamenteuse peut être fortement influencée par l'alimentation, par les variables physiologiques comme l'insuffisance rénale ou l'insuffisance hépatique, par l'âge du patient et également par les médicaments pris en co-médication (voir plus bas).
Influence de la fréquence d'administration
Les médicaments font l'objet, d'une manière très générale, d'administrations réitérées. Les objectifs à atteindre sont les mêmes que lors d'une administration unique :
- obtenir une efficacité thérapeutique dans les meilleurs délais ;
- maintenir en permanence une concentration plasmatique active dans l'intervalle thérapeutique ;
- éviter tout phénomène d'accumulation pouvant conduire à l'apparition d'effets toxiques.
Partant du principe de base que la dose choisie est bien adaptée, la fréquence d'administration est le facteur fondamental permettant de respecter les exigences énoncées. Les concentrations plasmatiques vont osciller entre deux valeurs, l'une maximale et l'autre minimale. Pour une efficacité optimale, la concentration maximale doit être inférieure au seuil toxique et la concentration minimale doit être supérieure au seuil thérapeutique.
Analyse non-compartimentale
L'analyse de données de pharmacocinétique ou la prédiction de propriétés pharmacocinétiques peut se faire à l'aide de méthodes compartimentales (qui divisent conceptuellement le corps en une série de boites homogènes) ou non-compartimentales. Les méthodes non-compartimentales estiment typiquement l'exposition à une substance sur la base de l'aire sous la courbe d'un graphique de l'évolution temporelle de la concentration de substance dans le sang.
Analyse compartimentale
Les méthodes compartimentales cherchent à décrire directement l'évolution temporelle simultanée des concentrations de la substance dans différentes parties du corps (organes, tissus ou cellules.) Ces méthodes sont complémentaires des méthodes expérimentales décrites ci-dessus en ce qu'elles permettent de mieux interpréter leurs résultats et de les extrapoler à des conditions non-observées ou même non-observables (par exemple chez la femme enceinte).
L'approche compartimentale consiste à décrire le corps comme un ensemble de boîtes (les compartiments), dans lesquelles la concentration est homogène. Le nombre de compartiments est variable : il peut être réduit à un unique compartiment (pour le modèle dit monocompartimental), mais n'est pas limité, selon les besoins de la modélisation. L'objectif est d'identifier un modèle, c'est-à-dire une fonction mathématique, permettant de décrire au mieux l'évolution des observations (concentrations plasmatiques) au cours du temps faisant suite à l'administration du médicament. Une fois cette fonction décrite, et si elle est suffisamment robuste, elle permettra d'identifier d'éventuelles sources de variabilité entre les individus et surtout de procéder à des simulations.
On différencie les modèles compartimentaux « classiques » et les modèles dits « physiologiques » (dits « modèles PBPK »), selon que ces compartiments prétendent décrire une réalité anatomique ou non. Ces derniers modèles s'appuient sur des données, le plus souvent obtenues in vitro, pour décrire des étapes pharmacocinétiques. Leur utilité dans le développement des médicaments fait actuellement l'objet de nombreuses études.
Variabilité Pharmacocinétique
La caractérisation du profil pharmacocinétique d'un médicament repose essentiellement sur les observations recueillies durant les phases cliniques de développement pharmaceutique. Ces essais conduits sur un petit échantillon de volontaires sains ou de patients sélectionnés selon des critères drastiques d'inclusion génèrent donc des paramètres moyens (Clairance, demi-vie d’élimination, volume de distribution, biodisponibilité) susceptibles de varier chez les individus en fonction de leur état physiopathologique ou de facteurs exogènes.
Parmi les principales causes de variabilité pharmacocinétique inter-individuelle, on retiendra:
- l'âge, qui se traduit par des modifications des fonctions digestives, d'une réduction des débits de perfusion des organes, d'une perte du débit de filtration glomérulaire, du perte des activités enzymatiques au niveau hépatique, d'une modification de l'albuminémie. - les polymorphismes génétiques touchant les gènes codant pour certaines isoformes enzymatiques impliquées dans la biotransformation de très nombreux médicaments (Cyp2D6, UGT1A1, DPD, Cyp2C9, Cyp2C19, CDA...), les gènes codant pour des transporteurs membranaires (familles ABC, MRP...), les gènes codant pour des facteurs transcriptionnels régulant l'expression d'enzymes ou de transporteurs (PXR, CAR). - les interactions médicamenteuses créant des phénomènes de compétition entre plusieurs médicaments envers un même système enzymatique ou un même transporteur, induisant des phénomènes d'induction/inhibition enzymatique, ou modifiant des variables biologiques (pH digestifs ou urinaires) affectant à leur tour les processus pharmacocinétiques. - les co-morbidités (états physio-pathologiques)touchant des organes impliqués dans les processus d'élimination et d'excrétion des xénobiotiques: sphère hépato-biliaire, fonction rénale pricnipalement.
Lorsqu'elle atteint un certain niveau, la variabilité pharmacocinétique peut conduite à une sortie de la zone thérapeutique à la posologie standard. Il convient alors de recalculer le schéma posologique afin de maintenir le patient dans une zone thérapeutique garantissant une efficacité pharmacologique sans risque de toxicité majeure. Une série limitée de classes pharmaco-thérapeutiques, mais représentant un très grand nombre de médicaments, est principalement concernée par cette problématique: anticancéreux, médicaments du système nerveux central, cardiologie, infectiologie.
Pharmacocinétique et Développement Pharmaceutique
Le développement d'une nouvelle entité chimique à visée thérapeutique (candidat-médicament) repose fondamentalement sur la caractérisation de son profil pharmacocinétique. De ce profil découlera: - le choix de la voie d'administration (forme orale, forme parentérale) - le choix d'un schéma posologique standard amenant une majorité de patients dans la zone thérapeutique requise pour exercer un effet thérapeutique sans entraîner de toxicités. - la compréhension des causes et facteurs possibles de variabilité pharmacocinétique: impact de l'âge, co-morbidités affectant les fonctions rénales et hépatiques, interactions médicamenteuses, polymorphismes génétiques... - l'identification des métabolites issus de la biotransformation hépatique et la caractérisation de leurs effets potentiels.
Il est admis que 40% des arrêts de développement pharmaceutique dans les années 90's étaient directement imputables à un profil pharmacocinétique défavorable (faible biodisponibilité orale, apparition de métabolites réactifs, biodistribution défavorable). Afin de palier ces écueils et réduire le taux d'attrition, l'industrie pharmaceutique intègre désormais une première phase exploratoire du profil pharmacocinétique en Discovery, afin d'éliminer rapidement les mauvais candidats selon la théorie du "quick kill". Cette stratégie a permis désormais de faire tomber à 10% le nombre d'arrêts de développement liés à un profil pharmacocinétique défavorable, en raison de l'élimination en phase de Recherche des molécules les moins intéressantes dans des études de Early DM-PK ("pharmacocinétique précoce") ou ADMET (ADME + hépatotoxicité).
Au delà des études précoces en phase de recherche, la pharmacocinétique est également étudiée in extenso:
- En développement non-clinique, sur des modèles in vitro et chez l'animal. - En développement clinique, chez le volontaire sain (études de phase I) ou le patient (études de phase I-II).
Notes et références
Articles connexes
- Paramètres pharmacocinétiques
- Phase biopharmaceutique
 Portail de la médecine
Portail de la médecine  Portail de la pharmacie
Portail de la pharmacie  Portail de la biochimie
Portail de la biochimie